
Bradley Williams, PhD, a budding physicist, began to feel his muscles weaken in 1981. In 2002, after years of progressive muscle wasting, he lost his ability to walk. Now, Williams needs help getting out of bed and doing other everyday tasks.
“It’s very challenging, and it can be hard to keep inspired to keep fighting,” said Williams.

Williams was diagnosed with a rare genetic disorder that was genetically confirmed in 2008. The disease called limb-girdle muscular dystrophy 2B (LGMD2B) is caused by the lack of a protein in the muscle membrane called dysferlin and therefore is a form of dysferlinopathy.
Dysferlin helps repair muscle membranes that undergo frequent damage due to physical activity and need constant upkeep. Normally, within seconds of an injury to the muscle membrane, dysferlin tethers lysosomal sacs to the muscle membrane, aiding in the release of an enzyme called acid sphingomyelinase (ASM) that is pivotal in the repair process. A lack of dysferlin reduces the injury-triggered release of ASM and compromises muscle repair.
“There still isn’t any treatment available, so those of us living with the disease have to accept its progressive nature and that we can’t do anything to slow it down,” said Williams. “A treatment is badly needed.”
A team of scientists at Children’s National Hospital, Washington, DC, has now adopted a new approach to develop a gene therapy for LGMD2B that circumvents the need for targeting hard-to-reach degenerating muscles with large doses and packaging the large dysferlin gene into a carrier vector—the two primary roadblocks in earlier and ongoing gene therapy efforts.
The preclinical study was published in an article in the Journal of Clinical Investigation on January 4 titled, “Secreted acid sphingomyelinase as a potential gene therapy for limb-girdle muscular dystrophy 2B“. It reports the first non-muscle targeted gene therapy for LGMD2B.
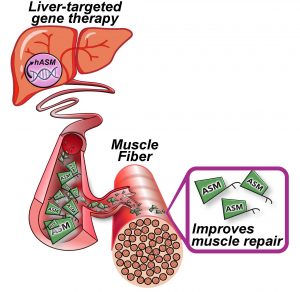
“This work is quite significant in carrying out a ‘compensatory protein therapy’ using secreted proteins from AAV-transduced liver as a means of effectively treating muscular dystrophy. The studies are done in an accepted mouse model of dysferlin deficiency for which there are no approved therapies, and no active clinical trials,” said Eric Hoffmann, PhD, professor and associate dean at Binghamton University and co-founder of ReveraGen, TRiNDS, and AGADA Biosciences, who was not involved in this study.
Williams, a self-taught expert on dysferlinopathy said, “The holdup on treatments is that the dysferlin gene doesn’t fit in an AAV, so it has to be delivered in two pieces. That means it’s not ‘low hanging fruit’ for companies developing gene therapy.”

“Our study provides proof-of-principle for a novel, non-muscle targeted gene therapy approach for the treatment of dysferlinopathy. This is the first liver-targeted LGMD2B therapy and first LGMD2B gene therapy that circumvents the need to package the large dysferlin gene,” said Jaiswal.
Hoffmann added, “This is a robust study that has important significance in terms of a new therapeutic approach for the LGMD2B type of muscular dystrophy, while also providing innovative data on the success of using the liver as a site of transgene production to compensate for skeletal muscle genetic deficiencies.”
The study measured the effectiveness of the treatment 12 weeks following the injection, but Jaiswal said, “based on the stabilization of the serum level of the ASM enzyme, we expect the benefits to be longer lasting.”
The authors reported that the treatment safely decreased muscle degeneration and increased muscle fiber size, resulting in increased muscle strength.
A common, early side effect linked with AAV therapy is liver toxicity. Jaiswal said, “We did not observe any overt damage in the liver, nor did we observe any serum indicator of liver toxicity. The animals maintained good health and maintained the expected growth in their body weight.”
One of the challenges in developing treatments for dysferlinopathies like LGMD2B has been the delivery of gene therapy vectors to all muscles in the body. “Muscles, which degenerate in this disease, are hard to reach, and thus require a large AAV dose. Liver targeting circumvents all these challenges,” said Jaiswal. Targeting the gene therapy to the liver allows its dissemination to all muscles body-wide through the circulation.

“Increased muscle degeneration necessitates greater muscle regeneration, and we found that improved repair of dysferlin-deficient myofibers by hASM-AAV reduces the need for regeneration, causing a two-fold decrease in the number of regenerated myofibers,” said Daniel Bittel, DPT, PhD, a postdoctoral fellow at the Center for Genetic Medicine Research at Children’s National and a first author of the study.

“Over and above the robust and significant findings in the LGMD2B model described in the manuscript, the studies may have more generalizable significance in that they describe a novel approach that may overcome some of the challenges that have been found in direct gene delivery to skeletal muscle—the very large target tissue (muscle constitutes nearly 30% of the body), the enormous amounts of gene therapy vectors that are required to target such a large target tissue, the remodeling of dystrophin muscle that may limit persistence of expression, and the large size of the gene causing many types of muscular dystrophy that do not fit into AAV vectors for direct gene therapy,” Hoffman said.
Jaiswal’s team is currently conducting long-term preclinical studies to test the durability and prolonged safety of this therapy. The researchers are also testing these benefits at different symptomatic stages to inform future clinical trials.
“It’s great to see this type of research identifying a potential treatment approach,” said Williams, who works for the Jain Foundation that focuses on LGMD2B research and has been living with the disease for the past forty years.




