Close at the heels of the FDA’s conditional approval the new drug for Alzheimer’s disease, aducanumab, that acts by reducing soluble and insoluble β amyloid, a new study led by scientists at the University of Cincinnati (UC) in collaboration with the Karolinska Institute in Sweden, sheds new light on the pathological mechanism underlying Alzheimer’s disease and a highly debated drug therapy that argues in favor of increasing soluble amyloid levels.
The study published in the journal EClinicalMedicine “High cerebrospinal amyloid-β42 is associated with normal cognition in individuals with brain amyloidosis” and funded by UC Gardner Neuroscience Institute, claims that the treatment of Alzheimer’s Disease might lie in increasing the levels of the soluble, fibril-producing, 42-amino acid, β amyloid peptide (Aβ42), that aggregates into neurotoxic plaques, tau tangles and instigates neuroinflammation—the hallmarks of the disease. Based on their evidence, the authors argue that in its original, soluble form Aβ42 is needed to keep the brain healthy.
“It’s not the plaques that are causing impaired cognition,” says Alberto Espay, PhD, the new study’s senior author and professor of neurology at UC and a member of the UC Gardner Neuroscience Institute. “Amyloid plaques are a consequence, not a cause [of Alzheimer’s disease],” says Espay.

Since the first identification of plaques in the brains of patients suffering from Alzheimer’s disease over a 100 years ago, Espay says scientists have focused on treatments to eliminate the plaques. But the UC team, he says, sees it differently. The hypothesis that Espay and his colleagues test in the paper is that the cognitive impairment in Alzheimer’s disease is due to a decrease in soluble β amyloid peptide and not due to the corresponding increase of insoluble β amyloid plaques.
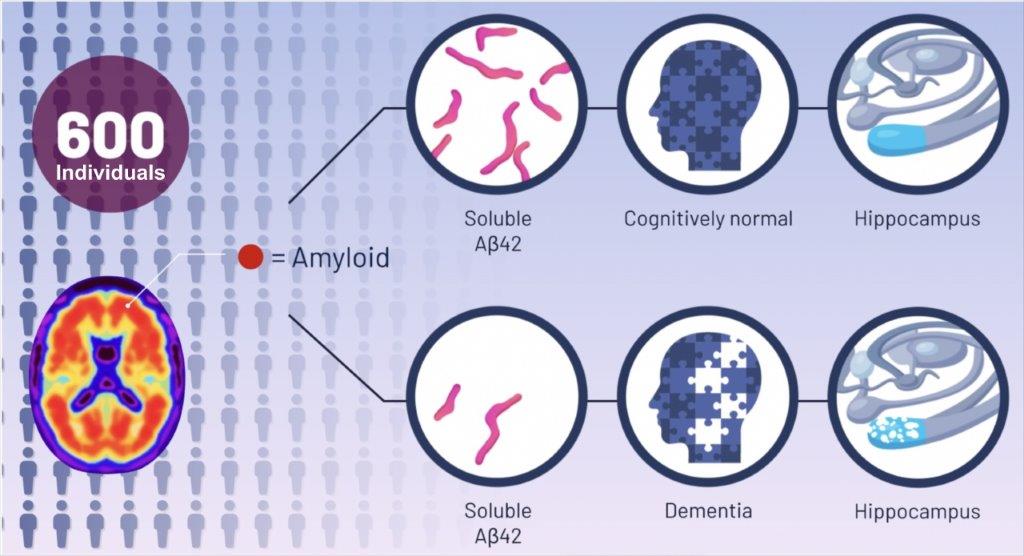
To test their hypothesis, the team analyzed the brain scans and cerebrospinal fluid from 598 participants in the Alzheimer’s Disease Neuroimaging Initiative study, who had amyloid plaques in their brains.
They compared the amounts of insoluble amyloid plaques and levels of soluble amyloid peptide in individuals with normal cognition, mild cognitive impairment and Alzheimer’s disease and found individuals with normal cognition had higher soluble amyloid levels (864.00 pg/ml) than individuals with mild cognitive impairment (768.60 pg/ml) or Alzheimer’s disease (617.46 pg/ml), despite the presence of brain amyloid plaques.
The researchers also found that higher levels of soluble amyloid β peptide were associated with a larger hippocampus, the area of the brain most important for memory.
Although by the age of 85, 60% of people will have these plaques, only 10% will develop dementia, the authors say.
“The key discovery from our analysis is that Alzheimer’s disease symptoms seem dependent on the depletion of the normal protein, which is in a soluble state, instead of when it aggregates into plaques,” says co-author Kariem Ezzat, PhD, scientist at the Karolinska Institute.
This leads the authors to conclude that the most relevant future therapeutic approach for Alzheimer’s disease will be replenishing soluble amyloid β peptide in the brain to their normal levels, says Espay. “Treatment,” says Espay, “may consist of increasing the soluble version of the protein in a manner that keeps the brain healthy while preventing the protein from hardening into plaques.”
The next step for the team is to test the findings and the novel treatment strategy in animal models. If successful, future treatments may overturn current approaches.
