

Co-founder, BioSkryb Genomics
The traditional view of human genetics is that we are born with a genome that provides a predictive model of our risks of developing different diseases. With a few exceptions, such as cancer, genetic testing has focused on identifying the genetic variations we inherited from our parents.
Recent studies have shown that the genomes in each of our cells are not static but are continuously changing because of the somatic mutations that occur as we develop and age. To more fully understand how those genetic changes contribute to human diseases, the models currently used to study genetic risk must be updated.
Mapping cellular evolution across our lifespan
Our cells rapidly divide and specialize after conception, transforming from a single cell into 37 trillion specialized cells that carefully orchestrate tissue function and health. This process, though, comes at a cost. Every cell division introduces uncorrected copying errors that leave permanent changes in the genomes of each of our cells. Furthermore, studies of human tissue have found that each cell in our body has about 100 new genetic changes when we are born. Since we possess trillions of cells when we are born, we begin our lives with enormous cellular genetic diversity that we have just begun characterizing.
We develop ever more genetic diversity as we age and become exposed to environmental mutagens, such as ultraviolet light and chemical mutagens. These factors, paired with the rate of cell division, impact the rate of somatic mutation acquisition across our tissues. Although single-cell genome sequencing is still young, most studies in the field have found that each of our cells acquires tens of new somatic mutations in cellular contexts each year of our lives.
With the development of more accurate methods of single-cell sequencing, we now know that those somatic mutations are present at higher levels than previously anticipated. The question has become: Are those mutations just passengers of life with little consequence, or do they contribute to the formation of human diseases?
Associating somatic changes with human diseases
With the capacity to accurately measure somatic mutations, we can now look at the roles these mutations play in a range of human diseases. It has been known that somatic mutations are associated with various skin conditions, which have been easy to identify based on distinct patterns that could be directly visualized by dermatologists. The development of next-generation sequencing has enabled us to identify causal somatic mutations for some of these conditions, such as Sturge-Weber Syndrome.
More recently, somatic mutations have been found in diseases of other tissues. For example, there have been extensive studies of clonal hematopoiesis of indeterminate potential after the identification of somatic mutations normally associated with the development of acute myelogenous leukemia in blood cells of elderly patients. Although these individuals have an elevated risk of developing leukemia, the greater concern is the increased risk of coronary artery and liver disease. This realization that somatic mutations in one tissue can have far-reaching consequences has spurred interest in studying these mutations in other disease contexts.
Somatic mutations also have been associated with both developmental anomalies, such as cortical dysplasia, as well as degenerative diseases associated with aging, such as Alzheimer’s. It has also been found that benign growths, such as vascular malformations, have specific somatic mutations that contribute to their formation. Further, other studies have shown that mutations in cells of the adaptive immune system can increase our risk of developing serious infections as we age.
This is just the beginning of our understanding of somatic mutations in human disease. The recent development of single-cell methods to identify these changes more accurately has spurred new interest in the field—we have only just begun to study somatic mutations in tissues and diseases on a large scale.
Discovering new views of cancer genomics
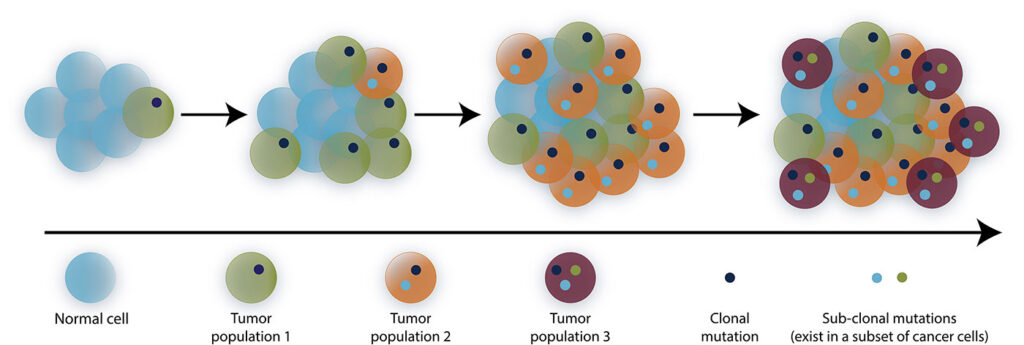
We have known for more than 40 years that somatic mutations contribute to cancer formation, but our understanding mostly comes from studies with tissue-level resolution. With single-cell sequencing tools, we now estimate that while each normal cell contains hundreds of somatic mutations, each cancer cell has from thousands to tens of thousands of these somatic changes. Further, those mutations differ across the tens to hundreds of billions of cells in each tumor.
This finding is transforming our view of cancer. Instead of a tissue with a limited number of genetic changes, we see a tissue composed of dynamic groups of related cells that also each harbor unique genetic changes and continuously co-evolve.
With further studies of cancer genomes in the single-cell context, we will develop a deeper understanding of the biology of cancer formation while providing new insights into clinically relevant outcomes—from high-resolution biomarkers of treatment resistance to new therapeutic targets to overcome that resistance.
Exploring implications of somatic mutation context
An important insight from early studies of somatic mosaicism is that most mutations do not cause human disease. Studies of known cancer genes in skin have found numerous small premalignant clones in each square centimeter of skin. However, the vast majority of those clones will never progress to cancer. This further supports the concept that a specific somatic mutation must occur in a specific cell type to result in the development of a disease.
In the context of human skin, for example, cancer-associated mutations may need to occur in a stem-like cell with the intrinsic capacity for self-renewal and other abilities that are required for cancer formation. This presents an opportunity to study somatic mutations in premalignant tissues to better predict which clones are most likely to transform to cancer so that we can intervene earlier and enable better patient outcomes.
Advancing single-cell solutions
Single-cell sequencing has enabled advancements in the field of somatic genetics over the past decade by providing the most quantifiable and accurate measurements of those mutations. Being able to accurately quantify the number, location, and signatures of somatic mutations in single cells has significant benefits over bulk tissue-based measurements that provide only a list of mutations without the cellular context. Moreover, methods that combine accurate mutation and phenotypic information from the same cells will enable scientists to begin studying how specific somatic mutations interact with that cellular context to produce normal and disease-associated cellular phenotypes.
We recently developed primary template-directed amplification (PTA), a method that can amplify the genomes of single cells much more accurately. It is allowing us to accurately call somatic mutations in almost the entire genome of a single cell for the first time. Furthermore, PTA in combination with whole transcriptome sequencing of the same cell allows us to study single-cell genomes in distinct cellular contexts.
These and other new methods are being supported by a new NIH Consortium, Somatic Mosaicism across Human Tissues (SMaHT), which aims to systematically study somatic mutations in a range of human tissues. These tools will undoubtedly be applied to study human diseases, including premalignant and malignant tissues.
With the accelerated adoption of more accurate single-cell technologies to study our evolving cellular genomes, scientists may soon conclude that, in addition to germline predisposition, somatic mutations are major drivers of human diseases that occur throughout our lives.
Charles Gawad, MD, PhD, is an associate professor of pediatrics at Stanford University. Gawad is also a co-founder of BioSkryb Genomics, where he is a member of the scientific advisory board and the board of directors.

