April 15, 2015 (Vol. 35, No. 8)
The Gene-Expression Undergrowth Have Been Well Trodden, but RNA Paths Want Wear, Too
A great deal of research on pathway analysis is currently focusing on RNA rather than proteins, and the complex RNA networks that regulate gene expression.
With the realization that more than 90% of the genome that is transcribed into RNA is not translated into protein, and the growing numbers of naturally occurring microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) being identified and characterized, the important role these RNAs play in normal biological processes and across human diseases is becoming increasingly clear.
This knowledge—combined with the available technology and strategies to decipher RNA pathways and link alterations in the levels or activity of miRNAs or lncRNAs to gene expression, epigenetic mechanisms, and protein activity in normal and disease phenotypes—is driving the development and clinical testing of novel drug targets and therapeutics that target regulatory RNAs.
For example, a microRNA was targeted in a Phase II clinical study that assessed the effect of miravirsen, an antisense oligonucleotide, in patients with hepatitis C. The study, which was described in 2013 in the New England Journal of Medicine, indicated that miravirsen sequesters the liver-specific microRNA miR-122 in a highly stable heteroduplex, thereby inhibiting its function.
Hepatitis C virus (HCV) depends on a functional interaction between its genome and miR-122 for viral stability and replication. According to the study, inhibition of miR-122 in HCV-infected patients was associated with decreased levels of HCV RNA that continued beyond the treatment period, without evidence of viral resistance.
The therapeutic potential of regulatory RNAs is also being assessed in other conditions such as cancer. Specifically, miRNAs and other ncRNAs in cancer initiation, progression, and metastasis are being studied by George Calin, M.D., Ph.D., a professor of experimental therapeutics, MD Anderson Cancer Center, University of Texas. Dr. Calin’s group is scouring the “microRNAome” to identify miRNAs of about 21–22 nucleotides that can serve as reliable biomarkers for cancer diagnosis and to guide decision-making in patient management, including as predictors of survival and response to drug therapy.
miRNAs are involved in every aspect of tumorigenesis, cancer progression, and dissemination. Not only are they expressed in tumor cells, they are also stably expressed in exosomes and are present in various bodily fluids, where they can act like hormones and signaling molecules. Comparative profiling of these fluids for differences in miRNA levels between patients with and without cancer could identify relevant biomarkers.

Hepatitis C virus depends on a functional interaction between its genome and miR-122 for viral stability and replication. Researchers recently used an antisense oligonucleotide that targets the liver-specific microRNA miR-122, blocking its function. [Bluebay2014/Fotolia]
Analyzing RNA Pathways
Dr. Calin and colleagues have described the significance of miRNA signatures obtained in recent studies involving miRNA profiling of human tumors. An overview appeared 2014 in CA: A Cancer Journal for Clinicians (“MicroRNAome genome: a treasure for cancer diagnosis and therapy”). Also, last February, Dr. Calin gave an account of his group’s work at the Molecular Med Tri Conference in San Francisco.
Technology is not holding back advances in the field of RNA pathway analysis according to Dr. Calin. The main bottleneck at present is in the design of prospective studies needed to confirm the predictive value of miRNA-based biomarkers.
Dr. Calin points to two other key challenges that scientists currently face in translating research findings into diagnostic, prognostic, and therapeutic tools. One is the difficulty in selecting an miRNA target, mainly because an individual miRNA could have a role in regulating tens, hundreds, or even thousands of protein-coding genes. For drug discovery, the aim is to identify miRNAs that affect a single pathway of interest to help limit off-target effects. The need for novel delivery systems for RNA-targeted drugs is another key challenge.
At the Molecular Med Tri Conference, Jean-Noel Billaud, Ph.D., principal scientist at Qiagen Bioinformatics, presented a case study demonstrating how the company’s Ingenuity Pathway Analysis technology can be used to conduct a systems biology analysis to identify the pathways, potential upstream regulators, and downstream outcomes involved in the host response to West Nile Virus (WNV) infection. Dr. Billaud also discussed how to interpret the results from a biological perspective.
In his presentation, Dr. Billaud described the first step in this analytical process as the acquisition of RNA sequence data using next-generation sequencing techniques for the purpose of characterizing and quantifying differential gene expression between an infected and uninfected cell. The CLC Cancer Research Workbench tool is used to process the sequence data, and the results are imported directly into the IPA system.
Analysis of differential gene expression aims to answer a series of key questions, including the following: What metabolic and/or signaling pathway(s) is activated or inhibited? Is there an overlap of the genes or pathways that are activated or inhibited? What are the potential upstream, downstream, functional, and phenotypic implications of this pathway activation or inhibition?
Dr. Billaud described other questions researchers might attempt to answer through the use of IPA: What are the identifying the underlying transcriptional programs? Which biological processes are involved and in what way? Are there splice variants of interest? What type of regulation is involved?
In the WNV case study, IPA predicted activation of the interferon signaling pathway and added statistically and functionally relevant biological processes to the WNV-related biochemical network the system developed. IPA is able to simulate the effects of interferon pathway activation on neighboring molecules and processes, which enables broader modeling of antiviral responses, prediction of the effects on viral replication, and identification of upstream transcriptional regulators of antiviral and related anti-inflammatory processes, for example.
These data and analytical capabilities may allow researchers to propose new hypotheses that connect molecules in regulatory networks to disease-related pathways in a predictive way, leading to the identification of a “master regulator” that could serve as a disease-specific drug target, according to Dr. Billaud.
In the WNV example, he described the use of the Molecule Activity Predictor (MAP) function in IPA to test the hypothesis that CLEC7A is a host susceptibility factor required by WNV to stimulate an immune response in the brains of infected patients, contributing to the development of life-threatening encephalitis. The MAP function simulates the inhibition or downregulation of CLEC7A, showing how it would likely reduce the risk of WNV-associated encephalitis. These types of hypotheses would then need to be tested and validated.
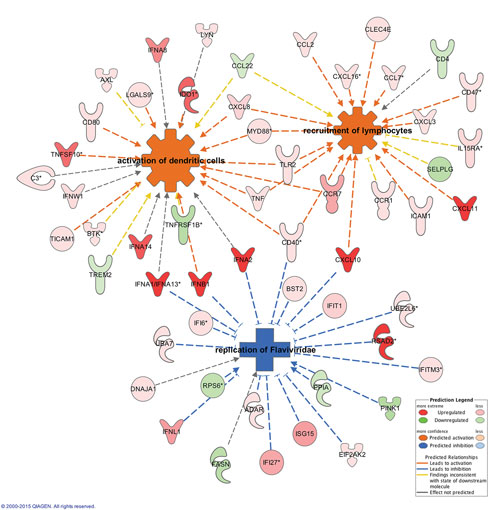
Using Qiagen’s Ingenuity Pathway Analysis, researchers can analyze relationships between molecules and diseases of interest by modeling how gene expression patterns affect functional outcomes or disease processes.
An Evolutionary Approach
In his laboratory at Hebrew University of Jerusalem, researcher Yuval Tabach, Ph.D., is using computational tools to analyze and compare the genomes and proteins of hundreds of species to identify evolutionary patterns of conservation and loss that point to connections between molecular pathways and disease.
“The main power of this phylogenetic profiling approach is that if you look at proteins across evolution, some are lost at certain points in certain species,” says Dr. Tabach. For example, proteins involved in the tricarboxylic acid (TCA) cycle have been highly conserved across some species, but have disappeared in others because those species have lost their mitochondria.
Dr. Tabach and colleagues have shown that sets of genes associated with particular diseases have similar phylogenetic profiles. They are also using this approach to identify genes associated with longevity, cancer resistance, and various extreme environmental conditions.
Phylogenetic profiling to connect patterns of conservation and loss across millions of years of evolution can be applied to entire proteins, protein domains, and RNA molecules such as microRNAs. The potential applicability of this approach to drug discovery and development is multifaceted.
For example, given a gene known to be related to a certain disease, the ability to identify other genes with a similar phylogenetic profile might reveal genetic factors that could explain incomplete penetrance or the variability of disease severity in different affected individuals. Alternatively, identification of a candidate gene in one patient could serve as the basis for identifying other key factors in other patients with the same disease using the phylogenetic profile.
Compared to strategies such as gene expression analysis or protein-protein interaction mapping for identifying disease-related genes, phylogenetic profiling “is much faster” and will become an increasingly powerful tool as the genome sequences of more species become available, explains Dr. Tabach.
The Israeli start-up company ReThink Pharmaceuticals is using the molecular networks generated through this phylogenetic profiling work for the purpose of drug repositioning. “If you know that a certain drug targets a gene, we can build a network to find other genes/proteins that interact with the drug target,” asserts Dr. Tabach, citing preliminary results that demonstrate the ability to predict additional effects of a drug candidate.
Pathways Driving B-Cell Differentiation
Robert C. Rickert, Ph.D., professor and director of the Tumor Microenvironment and Metastasis Program at Sanford-Burnham Medical Research Institute, is using conditional gene targeting to identify the genes and biochemical pathways that play a role at specific stages of B-cell differentiation. With this approach, it is possible to knock out targeted genes in a mouse at different stages of B-cell development, and to do so in an inducible fashion, allowing you “to look at how it affects different signal transduction pathways in a context-specific manner,” says Dr. Rickert.
When applied to a relevant mouse model of disease—such as a B-cell lymphoma—this inducible genetic system should yield effects similar to those that could be obtained with a drug capable of blocking the activity of the targeted gene product. Dr. Rickert and colleagues are exploring the similarity between the effects achieved with conditional gene targeting and those of recently approved drugs to treat chronic lymphocytic leukemia (CLL) and some forms of lymphoma such as idelalisib and ibrutinib, which are both inhibitors of the B-cell receptor pathway via blocking of PI3K or Bruton’s tyrosine kinase (BTK), respectively.
Dr. Rickert presented his group’s latest research at a Keystone Symposium Conference, PI 3-Kinase Signaling Pathways in Disease, which took place last January in Vancouver. In his talk, Dr. Rickert emphasized that the phosphatidyl inositol-3 kinase (PI3K) pathway is a major regulator B lymphocyte differentiation and function.
Dr. Rickert has also applied conditional gene targeting to compare the roles of the NFκB and PI3K pathways in B-cell maturation. He has shown that while both pathways are essential at some stages of B-cell differentiation, only one pathway may be necessary for B-cell maintenance and survival.
“Ultimately we want to gain more insight at the biochemical level into single cells and the heterogeneity of the cell populations we’re interested in,” says Dr. Rickert. Tumors and cancer cell populations are quite heterogeneic, and better biochemical tools are needed to be able to sort through these populations of cells and “look at some of the more interesting, rogue cells, such as cancer stem cells,” he adds.

