
One can hardly pick up a journal these days and not read about a novel CRISPR screen being used to delve into the mechanics of a model system. Rarely does a new technology so quickly take such a pervasive hold on biology. A major reason for this rapid adoption is the similarity in experimental design, approach, analysis, and reagents as those previously worked up for short hairpin RNA (shRNA) screens. Instead, the bottlenecks with CRISPR screens have been kicked down the road to the realms of the cellular context in which a given screen is applied.
A perfect example of how these cellular context restrictions may be overcome has been given by Sarah A. Teichmann’s laboratory, which recently presented a paper in Cell.1 Here, the cellular context is particularly challenging as it involves interrogating a “state” of primary murine T helper (Th) cells, in a system in which current CRISPR libraries do not function well.
The biological processes that govern the activation of naïve mouse Th cells and their differentiation into different effector subtypes (Th type 1 [Th1], Th2, Th17, and regulatory T cells [Tregs]) are intensely studied in immunology. However, with the exception of Th17,2 no one so far has successfully used an unbiased genetic perturbation screen in this context. Teichmann’s laboratory focused its efforts on the Th2 subtype, which play a major role in allergy and response to parasitic worm infections.
To study these cells, Teichmann’s laboratory had to overcome the potential showstopper that murine T cells are largely refractory to lentiviral vectors. Almost all available CRISPR libraries use lentiviruses as a delivery tool. Addgene, the popular plasmid repository, currently has 44 pooled libraries on offer, and with the exception of two plasmid libraries, all the pooled CRISPR libraries use a lentiviral vector to deliver the guide RNA (gRNA) library.3 To circumvent this, Teichmann and colleagues refactored a lentiviral-based mouse library4 into a retroviral system. This enabled the scientists to perform the first-ever large-scale CRISPR screen on mouse T cells.
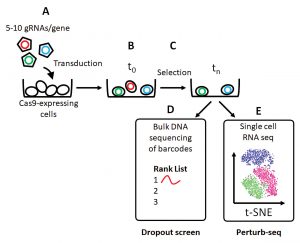
Another hurdle was to separate two distinct biological processes: activation of naïve Th cells to a state termed Th0, and differentiation of naïve Th cells to the Th2 subtype. To overcome this hurdle, Teichmann and colleagues worked in a manner analogous to one described in recent studies.5 They linked a traditional CRISPR dropout screen (Figure) to a phenotypic readout of Th2 function (assessed by flow cytometry–based reporter assays).
Like all functional screens, this CRISPR screen aimed to identify genes that influence a given phenotype in an unbiased fashion. Although not without technical challenges,6 CRISPR screens allow for the simultaneous interrogation of many genes in a high-throughput manner to uncover each gene’s contribution to a given phenotype.
Teichmann and colleagues could use their dropout screen in the Th0 arm to assess genes that are essential for “activation,” and then compare between Th0 and Th2 arms to assess genes that are essential for “differentiation.” By combining these data with RNA-seq for gene expression, ATAC-seq for chromatin accessibility, and ChIP-seq with key transcription factors, the scientists could generate a comprehensive resource describing the global genetic and epigenetic circuitry that governs Th2 cell activation and differentiation.
Such is the malleability of the CRISPR system that there are already CRISPR screens for many species and for almost every flavor of CRISPR technology. (Addgene currently offers knockout [n = 26], activation [n = 10], inhibition [n = 7], and barcoding [n = 1] libraries spanning five species.)
Combinatorial screens
While earlier CRISPR screens focused on benchmarking CRISPR, which is the new kid on the block, against shRNA, the prior gold standard,7,8 recent attention has focused on the benefits of combining different technologies rather than pitting them against each other.
So-called combinatorial screens bring an extra dimension and the opportunity to look for richer genetic interactions, such as cooperativity between genetic elements or synthetic lethality. These may involve combining more than one guide per cell9 or bringing orthologous CRISPR systems together10,11 to allow for both gene activation and knockout in the same cell.
Finally, by combining CRISPR screens with single-cell RNA-seq (scRNA-seq), the true power of the technology is beginning to become apparent. Though not at a genome-wide scale yet, technologies such as Perturb-seq,12,13 CRISPR-seq,14 and CROP-seq15 are enabling simultaneous detection of the effector gRNA and its consequence on the transcriptome in each individual cell, allowing for far more complex readouts than simple proliferation and reporter assays.
Technologies such as crisprQTL take this a step further, exploiting the vast amounts of genome-wide association study (GWAS) data to map enhancers with their targets.16 A fantastic example of the new wave of CRISPR screens is ECCITE-seq.17 Here, a CRISPR screen is combined with oligo-labeled antibodies, allowing the investigator to use scRNA-seq to read out the transcriptome, immune receptor clonotypes, cell-surface marker abundance, sample identity (to allow multiplexing), and gRNA identity. Looking ahead, as the cost of scRNA-seq continues to fall, and the computational challenges with analyzing such data are resolved, it won’t be long until these newer generations of screens reach genome scale.
If these technologies are to move into more complex model systems—for example, organoids or primary cells of limiting cellular abundance—we will once again have to match the appropriate technology with the cell type under study. In this instance, the current levels of library complexity (115,000 gRNAs on average for a mouse genome3) will have to be significantly reduced. This could occur by refactoring the gRNA designs to incorporate recent advances in indel prediction18,19 or by other means.
The right technology, the right cellular context—these two features are key in the realms of CRISPR screens.
Alasdair Russell, PhD, heads the Pre-Clinical Genome Editing Group, Cancer Research UK Cambridge Institute, University of Cambridge, U.K. Thanks to Tim Halim (CRUK Cambridge Institute, University of Cambridge, Cambridge, U.K.) and Ultan McDermott (AstraZeneca, Cambridge, U.K.) for insightful comments. This commentary is adapted with permission from The CRISPR Journal, where it was originally published.
References
- Henriksson J et al. Genome-wide CRISPR Screens in T Helper Cells Reveal Pervasive Crosstalk between Activation and Differentiation. Cell 2019; 176(4): 882–896. DOI:10.1016/j.cell.2018.11.044.
- Ciofani M et al. A validated regulatory network for Th17 cell specification. Cell 2012; 151, 289–303.
- LaManna C, Barrangou R. Enabling the rise of a CRISPR world. CRISPR J. 2018; 1: 205–208. DOI: 10.1089/crispr.2018.0022.
- Tzelepis K et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016; 17: 1193–1205.
- Li M et al. Genome-wide CRISPR-KO Screen Uncovers mTORC1-Mediated Gsk3 Regulation in Naive Pluripotency Maintenance and Dissolution. Cell Rep. 2018; 24: 489–502.
- Doench JG. Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet. 2018; 19: 67–80.
- Smith I et al. Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map. PLoS Biol. 2017; 15: 1–23.
- Morgens DW, Deans RM, Li A, Bassik MC. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol. 2016; 34: 634–636.
- Zhao D et al. Combinatorial CRISPR-Cas9 Metabolic Screens Reveal Critical Redox Control Points Dependent on the KEAP1-NRF2 Regulatory Axis. Mol. Cell 2018; 69: 648–663.
- Boettcher M et al. Dual gene activation and knockout screen reveals directional dependencies in genetic networks. Nat. Biotechnol. 2018; 36: 170–178.
- Najm FJ et al. Orthologous CRISPR-Cas9 enzymes for combinatorial genetic screens. Nat. Biotechnol. 2018; 36: 179–189.
- Dixit A et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 2016; 167: 1853–1866.
- Adamson B et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 2016; 167: 1867–1882.
- Jaitin DA et al. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 2016; 167: 1883–1896.
- Datlinger P et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods. 2017; 14: 297–301.
- Gasperini M et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 2019; 176: 377–390.
- Mimitou E et al. Expanding the CITE-seq tool-kit: Detection of proteins, transcriptomes, clonotypes and CRISPR perturbations with multiplexing, in a single assay. bioRxiv 2019. DOI: 10.1101/466466.
- Allen F et al. Mutations generated by repair of Cas9-induced double strand breaks are predictable from surrounding sequence. bioRxiv 2018; 37: 400341.
- Shen MW et al. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 2018; 563: 646–651. DOI: 10.1038/s41586-018-0686-x.
