
Excitement is palpable around emerging single-cell proteomics technologies that promise to parse the heterogeneity inherent in multicellular biological samples—a key limitation of conventional bulk sample experimentations—and arrive at relevant insights. Underlying techniques used to prepare large numbers of single-cell samples with adequate analyzable material in each, and acquire and interpret quality quantitative data, are continually evolving.
“Single-cell proteomics has transitioned from the shadows of skepticism to the forefront of research,” says Nikolai Slavov, PhD, director of the Single-Cell Proteomics Center at Northeastern University. “There are a lot of developments in the field, from high-throughput sample preparation to significant biological discoveries that are uniquely enabled by single-cell proteomics.”

Waters
Besides possibilities, single-cell proteomics poses challenges. This point is emphasized by Lee Gethings, PhD, a consulting scientist at Waters, and David Heywood, PhD, the company’s senior manager of biomedical research and global marketing. They say that sample prep, data acquisition, and data analysis are challenges shared by both single-cell and bulk sample analysis, whereas the amount of available material is the limiting factor for single-cell analysis. Consequently, in single-cell analysis it is critical to minimize losses and ensure that sufficient material is extracted from individual cells.
“An additional factor,” they point out, “is the ability to collect robust and reproducible quantitative data from large numbers of samples.” If this ability is to be maintained, high-throughput analyses must be available, ideally at an affordable cost.

The most common method that is used to arrive at single-cell samples is fluorescence-activated cell sorting (FACS). It uses the unique light-scattering attributes of fluorescently tagged cells to separate a heterogeneous mixture of cells, one cell at a time.
“FACS uses quite harsh conditions, and you never know in what condition your cell lands in the well or whether it lands in the well at all,” says Christoph Krisp, PhD, an application development scientist in Bruker’s proteomics team. “I like the CellenONE cell isolater from Cellenion. It gives us a visual of the single cell we are analyzing just before it is dispensed into the well.”
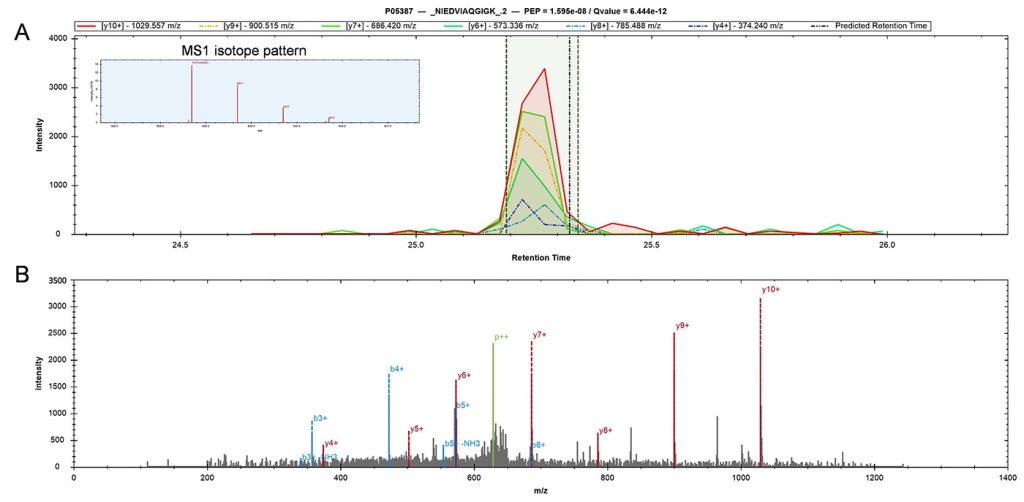
Related variations among proteins across single cells of the same type often reflect physiological states such as rest, cell division, intercellular communication, and resistance to drugs. Single-cell proteomics can quantify and interpret such covariation, provided the method used can achieve a high level of throughput and accuracy.
Minimizing losses
Slavov’s team has developed a flexible, automated, and parallelized method called nano-ProteOmic sample Preparation (nPOP). It simultaneously prepares 2,000 single-cell samples for mass spectrometry (MS) per batch. This involves lysing, digesting, and labeling individual cells in droplets as small as 8–20 nL on a fluorocarbon-coated glass slide.

Waters
Before proteins can be detected by MS, samples encounter plastic surfaces that adsorb proteins, which can be particularly detrimental for exceedingly small samples like single cells (<1 ng). According to Gethings and Heywood, strategies to minimize the effects of protein adsorption loss (PAL) range from the addition of specific detergents to implementing carrier protein methodologies.

Bruker
“Earlier, we thought that these plastic tubes had reduced [protein] binding capacity, but some proteins, particularly hydrophobic peptides, stick to these plastics,” Krisp notes. “We are now using a detergent [n-dodecyl-β-D-maltopyranoside, DDM] that prevents this binding. MS and detergents are not good friends, but this detergent is compatible with LC-MS [liquid chromatography-MS]. It helps recover most peptides from the vials.”
DDM is used at low enough concentrations to allow direct injection of samples into the column. It does not interfere with peptide ionization and elutes under high acetonitrile conditions during washouts.
Approaches orthogonal to single-cell proteomics, such as cytometry-based single-cell analysis (cyTOF) and in situ imaging, come with their own technical challenges. Gethings and Heywood note that in contrast to PAL, cyTOF encounters protein adsorption gain (PAG) through nonspecific antibody binding, which can be quantified and corrected. These scientists add that measurements generated from techniques using antibodies or aptamers require transformation, which can impact efficiency, selectivity, and availability.
Other experts do not find PAL to be a major challenge, primarily due to recent innovations in sample preparation. “Some people claim that protein adsorption loss is a huge problem, and of course it could be, with poor sample preparation, though the claims tend to be poorly supported by data,” Slavov remarks. “With nPOP, we see 95% efficiency. Based on these data and other experiments, my view is that protein adsorption loss is not a major bottleneck. Our biggest bottleneck is the relatively low efficiency of ionization by electrospray.”
Common buffers that contain sodium and phosphate salts reduce vapor pressure of droplets, lowering electrospray ionization. Volatile buffers such as ammonium acetate offer effective alternatives.
Labeling for coverage
Isobaric labels such as tandem mass tags (TMTs) are chemical tags that have the same mass when intact but reveal unique mass barcodes upon fragmentation. A major benefit of isobaric labels is that peptides from different samples appear as a single peak on MS spectra, which increases signal intensity, allowing simultaneous isolation of ions for multistage MS where peptides are identified and quantified. Slavov’s team showed isobaric carriers increase peptide identification without increasing the number of protein copies sampled from small samples.
“Isobaric labeling helps combine a lot of samples together to reduce MS time,” Krisp points out. “For single cells, it’s used to increase the number of peptides injected per experiment. We are also working on label-free strategies for faster gradients, but this always comes at the cost of quantitation accuracy.”
Isobaric labeling can generate impure spectra, lowering the accuracy of peptide detection. “Protein quantification with isobaric labels requires isolating and fragmenting one precursor at a time, which limits proteome coverage,” Slavov observes. “This can be mitigated with intelligent data acquisition.”
Even with existing MS instruments, it is possible, Slavov suggests, to leverage data acquisition and interpretation innovations to enhance peptide quantification 10-fold. Improvements in instrumentation, sample preparation, and peptide separation will further increase the coverage and quantitative accuracy of single-cell proteomics, while decreasing missing data.
Shotgun MS involves multidimensional separation of peptide mixes, MS/MS analysis, and identification through automated searches. Despite the method’s scalability, it remains biased toward abundant peptides in identifying and quantifying proteins.
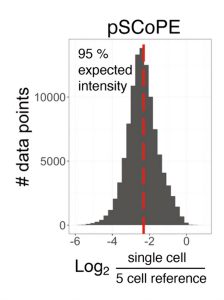
Compared to shotgun MS, prioritization results not only in twofold (or greater) increases in proteome coverage, but also in enhanced sensitivity and more complete data in single-cell protein analyses, by increasing the fraction of tandem MS scans assigned to peptide sequences, and the numbers of peptides per run and proteins quantified per cell. Prioritized single-cell proteomics (pSCoPE) analyzes thousands of prioritized peptides across single cells while analyzing identifiable peptides at full duty cycle, and it has been used to explore diverse polarized states of innate immune myeloid cells called macrophages.
Carrier proteome limits
Single-cell proteomics by MS benefits from isobaric labeling by spiking a carrier sample into a multiplexed experiment that is 10 to 200 times larger than the single-cell proteome. This “carrier proteome,” typically a mixture of cells that mimics experimental samples, facilitates peptide selection and identification. The concept is akin to identifying a blade of grass by holding it next to a tree to avoid missing the grass due to its inconspicuousness. However, it is unclear how accurately a single-cell signal could be measured against an overwhelming carrier signal.
“It’s best to use the same cells you want to analyze, as your carrier proteome,” Krisp advises. “If you use cheap, bulk, commercially available digests of cell lines, they may not completely reflect the cells you are looking at because of differences in culturing or processing.”
The carrier proteome, used as a reference to compare single-cell signals, can limit ionization efficiencies.
“Let’s say you have something of huge abundance compared to something really small,” Krisp suggests. “Even if you adequately fragment your peptides, you may be unable to see the reporter ions. The reporter ions from your booster channel may take up the entire space in the accumulator.”
Research led by Bernhard Küster, PhD, professor of bioanalytics at the Technical University of Munich, and Christopher Rose, PhD, director of microchemistry, proteomics, and lipidomics at Genentech, demonstrates that an increase in carrier proteome requires a parallel increase in the number of ions sampled to maintain quantitative accuracy.
Multiplexing for throughput
Single-cell proteomes can either be analyzed one label-free cell at time or multiplexed using isobaric or non-isobaric labels for expediency. “The [proteomics] community is increasingly focusing on approaches to increase throughput and collect larger datasets of both cell lines and primary samples,” Slavov relates. “Higher throughput is enabled by using shorter gradients and multiplexing samples.”
Multiplexing of samples and peptide analysis has advanced with plexDIA, an experimental and computational framework developed in Slavov’s laboratory. Whereas existing methods are suitable for achieving high throughput and depth in large samples, they are not suitable for low sample amounts. In its current version, plexDIA uses three-plex non-isobaric mass tags and one-hour active gradients to quantify three times more protein ratios in nanogram amounts of samples. In individual human cells, plexDIA can quantify over 1,000 proteins per cell and achieve 98% data completeness, establishing an enhanced framework for high-throughput, sensitive, and quantitative proteomics.
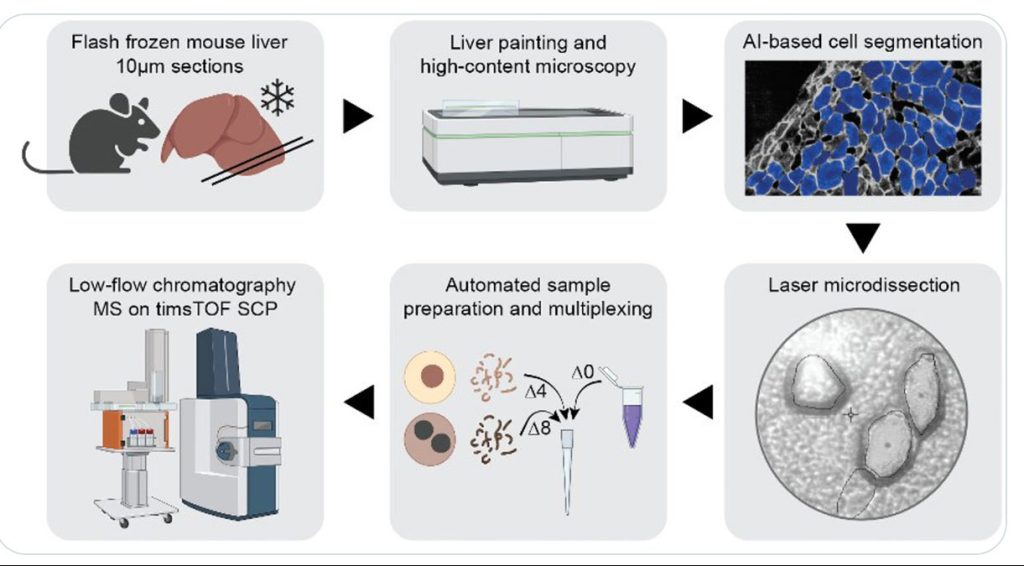
Single-cell proteomics using MS is not limited to cell culture samples. A team led by Matthias Mann, PhD, director and scientific member of the Max Planck Institute of Biochemistry, has integrated high-content imaging, laser microdissection, and multiplexed MS to develop a new technology—single-cell Deep Visual Proteomics (scDVP). The team has applied the new technology to spatially map the mouse liver proteome at a depth of 1,700 proteins from individual cells and trained machine learning algorithms to infer spatial proteomes from imaging data.
Next-generation approaches
Although DNA is amplifiable, proteins are not. This is a major obstacle for proteomics technologies. Next-generation multiomics systems built for scale, speed, sensitivity, and “interactome” detection work around this limitation through aptamer recognizers in targeted panels or molecule-agnostic sequencing, says Natasha Wagner, product manager, Encodia. “Recognizers” with DNA barcode tags—affinity reagent moieties that latch on to amino-terminal residues and identify each peptide-DNA chimera—have been key in developing next-generation technologies.
“Biology and how it interacts with its environment is not a one ‘ome’ problem,” insists Nigel P. Beard, PhD, chief technology officer, Encodia. “At the moment, proteome is the laggard. It’s not the fault of MS, which was never built to resolve proteins at this scale. We’re about to add a complementary toolkit to the mix.” Encodia is developing ProteoCode, a technology that involves “reverse translating” protein sequences into a DNA library. This would allow single-molecule identification and quantification at unprecedented scales and costs.
“MS is the gold standard of proteomics, but we are using next-generation sequencing as a readout,” Beard notes. “Our technical hurdles involve achieving Edman-like degradation to digest an immobilized peptide backbone, one amino acid at a time, without harming the writing of a DNA barcode that is building up at the same time.”
No Swiss army-knife approach exists to uncover the secrets of the proteome at single-cell resolution. Technologies, however disparate in principle and protocol, will ultimately cross-pollinate and complement each other to provide unique profiles of the proteome that when pieced together will offer a richer, more panoramic view.