
The ability to analyze the levels of gene expression within a single-cell has revolutionized the understanding of biology in fields from cancer to neuroscience. Since the first single-cell RNA-sequencing (scRNA-seq) study was published over a decade ago, it has proved a valuable technology; the information gleaned has aided in revealing heterogeneity among cell populations, identifying new cell types, differentiating cellular subtypes, and uncovering rare cells.
Some research questions require a deeper understanding beyond the gene expression within a single cell. For example, the ability to uncover the epigenetic profile of a cell in order to capture open chromatin regions throughout the genome, allowing researchers to identify which noncoding sequences may be important for turning on and off genes. To do that, ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) is a tool that can be implemented.
The power of ATAC-seq, explained Ben Hindson, PhD, chief scientific officer of 10X Genomics, is that you do not need to decide ahead of time what transcription factors might be important for your experiment. ATAC-seq allows the profiling of all accessible chromatin regions. Using known transcription factor sequence motifs, software computationally scans the genome for transcription factors enriched in accessible chromatin regions, allowing the researcher to “pull out transcription factors that may be important for a specific population of cells, and also defining the chromatin regions the transcription factors may bind.”
The ability to simultaneously measure ATAC and RNA-seq in the same cell would allow the understanding of the epigenetic factors that control the expression of genes with single-cell resolution, noted Ansuman Satpathy, MD, PhD, assistant professor of pathology, Stanford University School of Medicine. Rather than taking each measurement separately and inferring their linkage through computational methods, this method allows the direct capture of this information in each individual cell.
Up until now, nobody has been able to bring the ability to query both pieces of biology in the same cell into the market. But 10X Genomics Chromium Single Cell Multiome ATAC + Gene Expression—first previewed in February at the AGBT meeting in Marco Island, FL, and being released today—is capable of simultaneously profiling the epigenome and transcriptome simultaneously from the same single cell.
“Until now, we have relied on computational prediction to match a cell’s epigenome to a single-cell gene expression profile,” said Holger Heyn, PhD, team leader at the National Centre for Genomic Analysis at Spain’s Centro Nacional de Análisis Genómico, and who is working on delineating the dynamics underlying B-cell differentiation and activation.
Indeed, 10X Genomics sells separate RNA-seq kits and a scATAC-seq kit. But they could not simply merge the two kits together in order to combine to two modalities. The new kit that combines both techniques uses new gel beads and chemistry that enable researchers to barcode both mRNA and DNA fragments. After sequencing, the barcodes are used to pair the gene expression and chromatin profiles back together, from the same single cell. “mRNA tends to be less stable than DNA,” explained Hindson, “so a large part of the effort was making sure the assay could capture both mRNA and DNA from cells, at high sensitivity, and be reflective of underlying biology.” There is also a newly designed software analysis pipeline to leverage the two different data types.
The workflow is approximately two days, or one day longer than the scATAC-only version. Because researchers are making two NGS libraries (ATAC and RNA) from the same starting sample, it takes more hands-on time to complete.
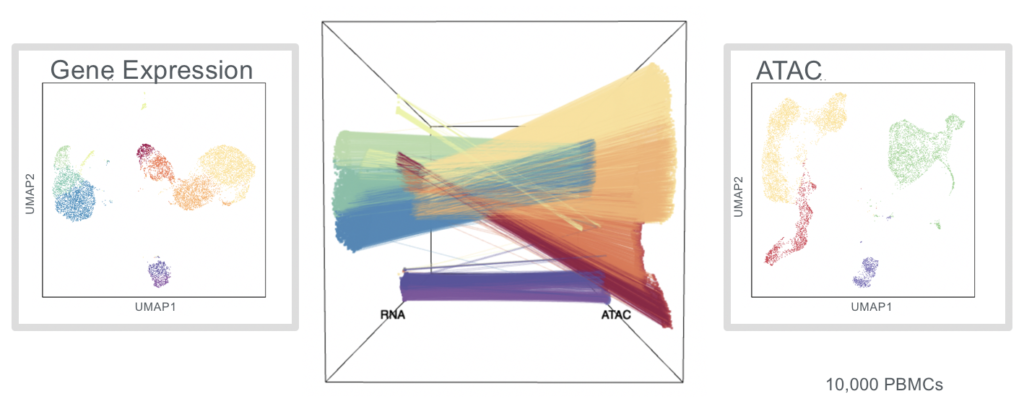
The ability to combine epigenetic and transcriptomic data from a single cell could appeal to researchers in varied fields, including oncology, immunology, neuroscience, and stem cell and developmental biology.
For example, a major barrier for effective immunotherapies, explained Satpathy, is the development of intratumoral T-cell exhaustion. Satpathy’s lab had previously mapped scRNA-seq and scATAC-seq profiles in human exhaustion, but are now focusing on a way to link these two together to understand the gene regulatory mechanisms that control dysfunctional T cell gene expression in each cell.
Satpathy expects that the integration of both types of information will provide a deeper understanding of the types of cells that exist in complex tissues, and their molecular similarities to one another. For example, the information could be used to more easily connect the differentiation trajectories of stem and progenitor cells to their terminal daughter cells, and to understand the precise regulation of each step in this process. Similarly, in settings of disease, it will allow them to be able to more comprehensively define the precise molecular switches that control a disease-associated or “dysfunctional” cell type, compared to its healthy counterpart.
In addition, Satpathy told GEN that measuring these principles in primary human tumors reveals insights that cannot be obtained in animal models. To that end, he will employ the new 10X solution on primary patient samples. This method will be particularly useful, he asserted, for low-material samples, for example, clinical specimens. When dealing with these types of samples, they would like to obtain both measurements, but often have to choose one or the other due to sample constraints.
Neuroscience researchers are also making a plan to capture epigenomic and transcriptomic data from a single cell. Panagiotis Roussos, MD, PhD, associate professor of genetics and genomics sciences at the Icahn School of Medicine at Mount Sinai, noted that this product will allow them to “better understand the mechanisms affected by the noncoding risk genetic variation across a wide range of neuropsychiatric diseases, including Alzheimer’s, Parkinson’s, Schizophrenia, bipolar disorder, and major depression, along with different severity of neuropathology and clinical symptomatology.”





