
After notching a handful of regulatory approvals in the last four years, chimeric antigen receptor (CAR) T-cell therapies are beginning to deliver remarkable efficacy for cancer patients—albeit with some concrete limitations. Tapping into a range of new technologies, biotechnology companies are developing the next generation of cell therapies intended to broaden the range of treatable cancers, reduce toxicities, and improve patient access.
CAR T cells are engineered cancer immunotherapies that boost or hijack a person’s immune system to target their cancer. To date, all of the approved CAR T-cell therapies are autologous approaches—dependent on extracting a patient’s T cells, engineering them to express a CAR that redirects them to tumor targets, expanding them ex vivo, and reintroducing them to the patient.
The manufacturing process is complex, time consuming, and expensive. Moreover, no CAR T-cell therapies have been approved yet for patients with solid tumors—the most common cancers—and they can cause a range of serious-to-fatal inflammatory and immune toxicities that have prevented them from being deployed before other avenues have been exhausted.
Companies and researchers are testing a range of new methods to abrogate these limitations: developing off-the-shelf therapies, better-targeting CARs, and even therapies that remodel tumors themselves for better CAR T-cell infiltration.
Starting from scratch
Because cancer is such a dynamic disease—with differences between patients and cancer types, and even between (and within) tumors—treatment courses are becoming increasingly personalized. It’s a lengthy and expensive approach, so many cell therapy companies are moving in two tracks: developing autologous therapies while working in parallel to develop the technologies to eventually enable allogeneic, or off-the-shelf, therapies.
Working sequentially this way makes sense as a corporate de-risking strategy, but it may ultimately be irrelevant. Several biotechnology companies are using induced pluripotent stem cells (iPSCs) that can serve as precursors to T cells and that can be edited ex vivo to carry a CAR and coaxed into forming T cells in cell banks. Fate Therapeutics received the first U.S. Food and Drug Administration (FDA) clearance last year to begin clinical trials to evaluate an iPSC-derived CAR T-cell therapy for FT819. It targets CD19 expressed by certain hematological B-cell malignancies.
The approach is much more efficient than working with donor-derived cells, says Greg Fiore, MD, CEO of iPSC player Exacis Biotherapeutics. According to Fiore, in autologous approaches, donor-derived T cells need to be screened after adding genes because only a small percentage of cells will take up the gene therapy used and properly display the CARs. “We don’t have any of those challenges,” Fiore asserts, “because we’re editing at the iPSC level.”
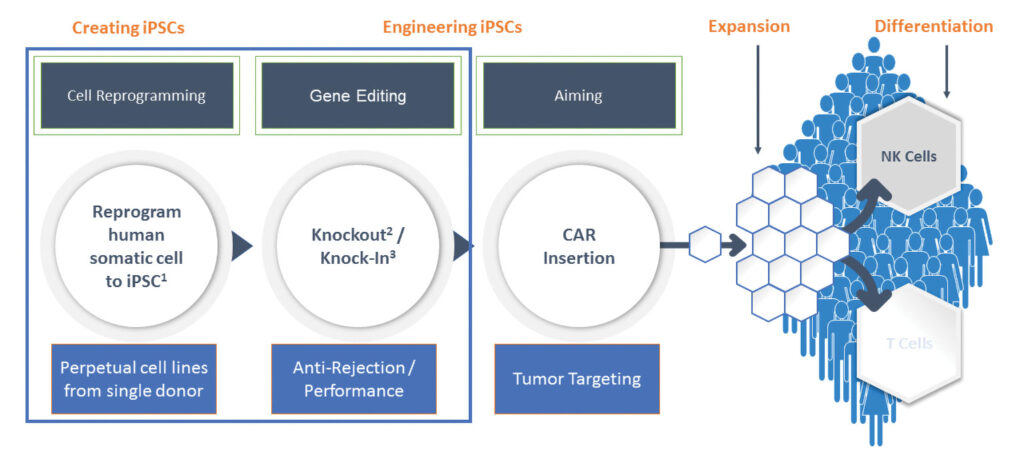
According to Fiore, fully edited iPSCs can be clonally expanded faster than T cells, and Exacis’s mRNA approach to cell reprogramming is more efficient than DNA approaches with viral vectors.
Rejecting rejection
Immune systems are built to reject foreign cells, which can contribute to CAR T-cell toxicity and a lack of cell persistence (Sterner RC, Sterner RM. Blood Cancer J. 2021; 11: 69). To circumvent this, Exacis is attempting to cloak its CAR T cells by knocking out a gene that encodes for a major histocompatibility complex (MHC) protein. MHCs bind peptide fragments in the cell, such as those from infecting pathogens or cancerous mutations, and display them on cell surfaces to make them identifiable to the immune system, triggering an adaptive immune response.
But removing this “fingerprint” from cells is just the first hurdle of avoiding rejection, says Sonja Schrepfer, MD, PhD, scientific founder, senior vice president, and head of Hypoimmune Platform, Sana Biotechnology. Because viruses also downregulate MHCs to avoid detection during infection, the immune system has a secondary defense system, where natural killer cells and macrophages attack and clear cells missing the identifiers, including foreign T cells. “That’s the biggest hurdle,” Schrepfer points out.
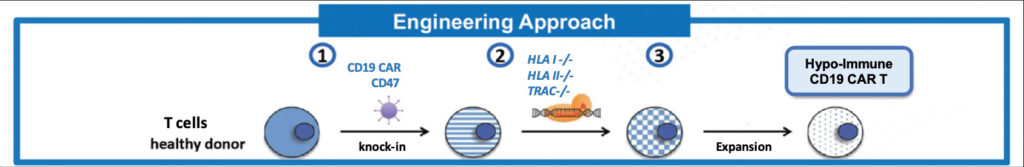
Sana has generated huge investor excitement, closing a $675.6 million initial public offering last February. The company is engineering hypoimmune cells that overexpress CD47, a surface protein known to send a “don’t eat me” signal to macrophages. Last June, at the annual meeting of the International Society for Stem Cell Research, Sana presented data from a study in nonhuman primates with healthy immune systems showing hypoimmune iPSCs were durable, surviving at least four months with no systemic immune response detected. By comparison, unmodified iPSCs quickly activated T cells and were cleared.
According to Schrepfer, “The nonhuman primate model is a very high bar, because in this model, the immune system is very reactive.” The company is aiming to launch its first clinical trials next year with two CAR T-cell programs. Sana’s lead allogeneic, ex vivo–engineered candidate is SC291, which incorporates the company’s hypoimmune technology and targets CD19—the same antigen that is targeted in most approved CAR T-cell therapies.
Bringing the outside, inside
Sana is also developing a second CAR T-cell platform using a gene therapy approach. There, the cell engineering will occur inside the body, via fusogen delivery vehicles that bind to targeted T-cell surface proteins. Steve Harr, MD, Sana’s president and CEO, says the lead fusogen CAR T cell—also targeting CD19—is on pace for human testing next year as well.
Sana is one of several companies pursuing an in vivo approach. The appeals are clear. First, taking an in vivo approach allows developers to sidestep the manufacturing hurdles that must be cleared along ex vivo approaches. Second, hijacking the body’s own T cells allows developers to avoid the problem of immune rejection altogether.
A gene therapy approach is also being explored by Umoja Biopharma. Fresh off a $210 million Series B financing round in June, Umoja is racing toward the clinic, on track for next year with its lead ex vivo CAR T-cell program, UB-TT170. This program employs the company’s TumorTag platform, which uses small-molecule bispecific adapters to tag folate-expressing osteosarcoma tumors and their stroma. Once tagging is accomplished, autologous CAR T cells are delivered that target the tags.
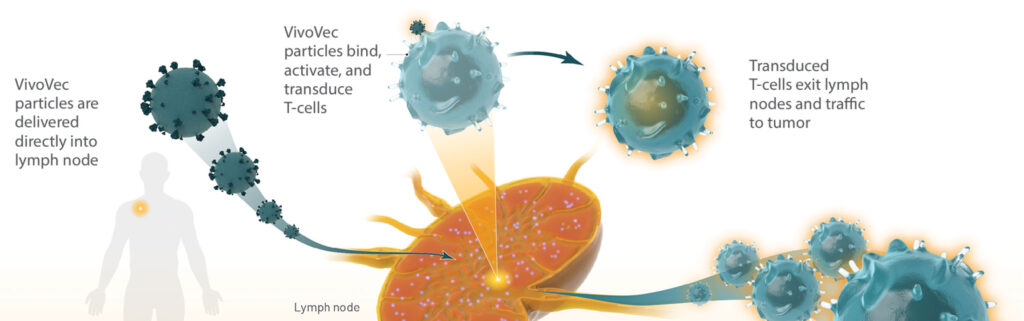
The TumorTag platform is just the first piece of Umoja’s approach. Ultimately, the company plans to combine the technology with its in vivo CAR T-cell platform, engineered using a lentivirus-based gene modifying agent.
Developing an in vivo platform meant creating preclinical models that are more complex than those used for ex vivo therapies. “Everyone that’s in this space is faced with the challenge that the immune system has evolved to be exceptionally unique—across species, across individuals,” says Shon Green, PhD, senior director of translational sciences at Umoha. “Most of the things you’re trying to develop bind very specific human targets, and also rely on very unique interactions within the same host.”
For ex vivo therapies, platforms are often tested with a xenograft tumor in an immune-compromised mouse model. But for a therapy designed to modify human immune cells in vivo, Umoja needs mice engrafted with a rudimentary human immune system.
“We’re trying to create a mouse model where there isn’t a lot of xenogenic or allogeneic activation of T cells,” Green explains. “[We want] a really accurate picture of how well our product will work in a human, in terms of activating and transducing those T cells.”
A model in development uses immunodeficient mice with MHC proteins knocked out, engrafted with mature peripheral blood mononuclear cells. The company is working on transplanting human tumors that also have MHC proteins knocked out. According to Green, they resemble, as much as possible, what Umoja’s engineered virus particles will encounter in humans.
Expanding possibilities
The industry is also very interested in expanding the capabilities of CAR T cells against other targets and other cancer types. Myrio Therapeutics, for example, is developing an antibody fragment library focused on peptide-MHC complexes, which can be used to construct CARs against challenging targets.
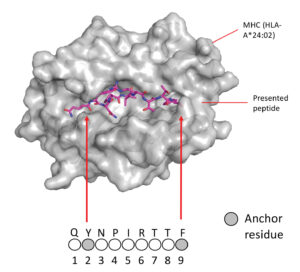
At the forthcoming CAR-TCR Summit 2021, Myrio CEO Pete Smith, PhD, will present data from a collaboration with researchers at Children’s Hospital of Philadelphia. The researchers developed a CAR T cell against PHOX2B, a novel therapeutic target in neuroblastoma, and tested it in a patient-derived mouse model. “The CAR T cell led to 100% tumor eradication,” Smith says. By comparison, in the same model, CD19-targeted CAR T cells and unmodified T cells had no effect. He adds that similar activity has been shown on multiple animal models.
Finding novel solid tumor antigens to target is an important piece of the challenge, says Yi Fan, MD, PhD, an associate professor of radiation oncology at University of Pennsylvania. But given the number of strategies companies are exploring, he’s optimistic that enough targets could be identified in the next few years to help put the more widespread cancers in play.
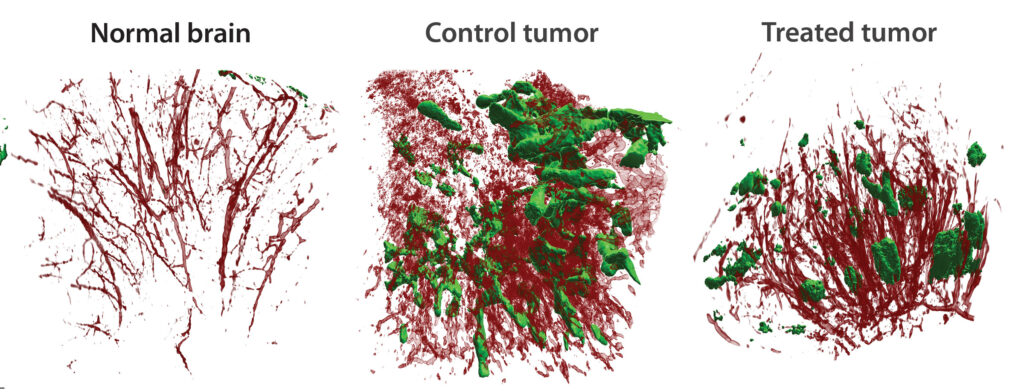
A bigger challenge is penetrating the solid tumor microenvironment. “There are a lot of obstacles, like the stroma and the vasculature,” Fan notes. In the clinic, patients may receive millions of CAR T cells in a treatment, but only 1–2% make it into a solid tumor. “And even if they do,” he adds, “they do not become active.”
In a paper published last November (Ma et al. Nature Cancer 2021; 2: 83–97), Fan’s team identified PAK4, a protein kinase that regulates aberrant vascularization. “Using a pharmacological inhibitor, we found that we can reprogram the vasculature into a beautiful, treelike structure,” he reports, “and [that this structure can allow] more T cells to get in.”
Adhesion proteins in the vasculature improve T-cell recruitment to tumors—otherwise, the T cells slide by as if they were passing through a polished pipe, Fan remarks. PAK4 inhibition increased the expression of these adhesion proteins in the vasculature, allowing the T cells to infiltrate the tumor. Fan’s team is planning a clinical trial to see if a PAK4 inhibitor can boost CAR T-cell efficacy in patients with solid cancers.
The tumor is also protected from T cells by immunosuppressive macrophages inside the microenvironment, which researchers have shown can be reprogrammed (Oyarce et al. Oncoimmunology 2021; 10: 1898753) into a tumor-killing phenotype. Fan’s group is exploring this approach to boost CAR T-cell therapy as well, and he is launching Radix Therapeutics to develop immunotherapies focused on the solid tumor microenvironment. He observes that developers in the field are justifiably excited by T cells, but he adds that T cells can’t kill tumors if they can’t reach them.