
Post-translational modifications (PTMs)—the chemical changes that proteins undergo following biosynthesis—account for most protein forms, or proteoforms. Indeed, according to conservative estimates, there are 1 million proteoforms, 90% of which are believed to be PTM-derived proteoforms. Estimates for the number of proteoforms and the percentage of PTM-derived proteoforms sometimes range quite a bit higher, depending on how the proteoforms are defined.1
The many PTM-derived proteoforms are important because they add greatly to the functional diversity of the proteome. They correspond to dynamic, labile, and often cell- or tissue-specific PTMs that regulate cellular pathways. Presumably, these PTMs have much to tell researchers in the life sciences. But these researchers will have to listen closely. Many PTMs are rare, subtle, and fleeting.
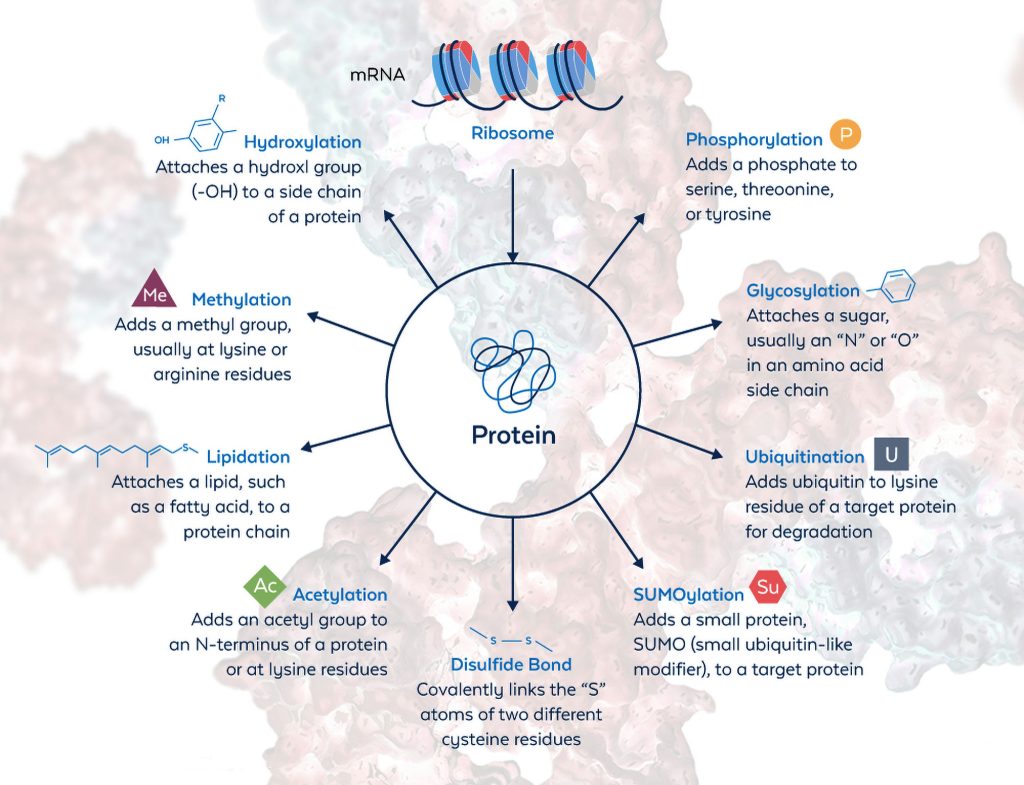
A more fundamental issue is that there are many types of PTMs. Indeed, there are over 400. About 90% of PTMs, however, are of just three types—phosphorylation, acetylation, and ubiquitination. Other major PTM types include glycosylation, succinylation, methylation, malonylation, SUMOylation, palmitoylation, myristoylation, prenylation, and sulfation.2 PTMs can be simple or complex. Examples of the latter include glycosylation and polyubiquitination. These PTMs may result in linear-chain and branched-chain structures.
Although the proteome is vast, it needn’t overwhelm. And although it is full of intricacy, it needn’t bewilder. The proteome may be studied with an expanding array of tools and technologies. Many of these tools and technologies are described in this article.
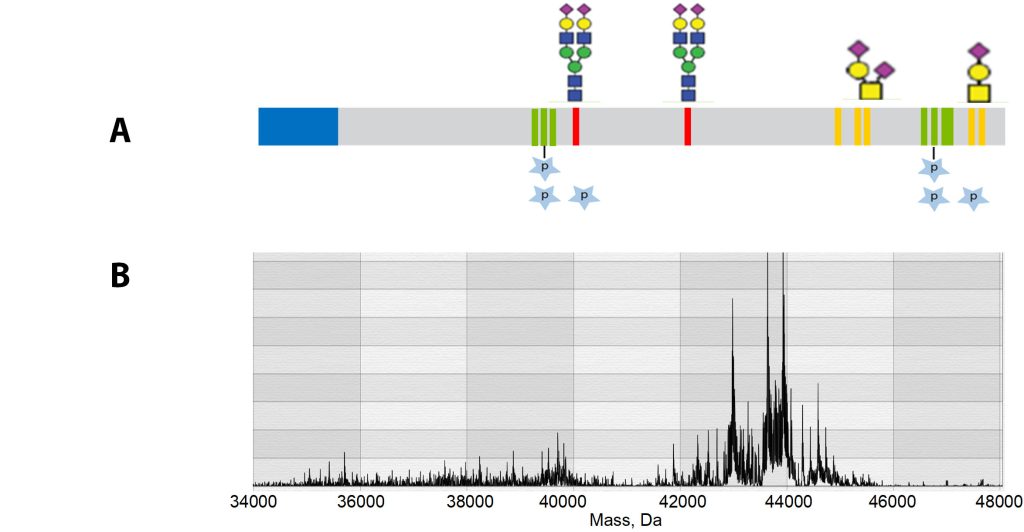
Accelerating proteoform discovery
Aaron M. Robitaille, PhD, director of marketing, mass spectrometry, Thermo Fisher Scientific, notes that the first PTM was discovered over 100 years ago.3 He adds that it is only now that we are starting to appreciate how many proteoforms exist.
Typically, PTMs are analyzed in both temporal and spatial terms to clarify how they regulate cellular function. According to Robitaille, a classic example is phosphorylation, where the addition or subtraction of phosphate to an amino acid can activate or deactivate a protein. This PTM can serve as a kind of on/off switch.
The sample enrichment requirement prior to analysis to increase detection of low-abundance proteins and/or to reduce sample complexity is a key challenge. “Proteoforms can be transient and rare,” Robitaille notes. “They also can become dysregulated in disease and thus are important therapeutic targets. But small clinical samples such as biopsies present a limitation for enrichment.”
According to Robitaille, mass spectrometry (MS) offers the best sensitivity and specificity to study PTMs. He notes that once a standardized MS assay is developed, it can be deployed globally in multiple settings and produce the same quantitative results.
Robitaille indicates that the generation of Selective Temporal Overview of Resonant Ions (STORI) plots4 can be facilitated by an MS solution called Thermo Scientific Direct Mass Technology mode. Moreover, it can enable precise measurements of heavily modified proteoforms, such as those produced from glycosylation processes that are too complex to resolve using customary methods.
Traditionally, affinity reagents such as antibodies or aptamers must be generated for each specific PTM modification site and verified for specificity, requiring enormous amounts of reagents. In contrast, high-resolution accurate-mass (HRAM) instrumentation enables the characterization of proteoforms in their naïve state as well as the discovery of new PTMs with specific site localization. This fit-for-purpose MS technology is designed specifically to help scale to the level of 6 million proteoforms.
“We are analyzing single proteoforms without the need for enrichment,” Robitaille emphasizes. “The incredible sensitivity shows the heterogeneity of the biological system.” In the future, a range of MS-based assays including enzymatic activity, thermal shift, and protein degradation (PROTAC) studies will potentially address the functional effect of PTMs on protein-based activity.
“MS instruments do not directly measure function,” Robitaille points out. “You need a properly controlled experimental design that can take advantage of the sensitive and specific MS readout to understand function. Many PTMs are understudied due to a lack of enrichment tools or affinity reagents, representing a key area where MS empowered by Thermo Scientific Orbitrap technology and Direct Mass Technology mode can play a major role.”
Grappling with glycobiology
The rise of therapeutic antibodies results in the need to characterize lot-to-lot glycan profiles. “It is well known how glycans influence antibodies’ therapeutic potency,” says Anthony Person, PhD, senior director, Protein Business Unit, Bio-Techne. For example, certain immunoglobulin G1 antibodies rely on the Fc-mediated immune effector function, known as antibody-dependent cellular cytotoxicity (ADCC), as a central way to deplete tumor cells. Core fucosylation on N-glycans on antibody heavy chains directly influences the binding affinity of the antibody’s Fc portion to the FcgRIIIa receptor present on immune effector cells like natural killer cells. Generally, antibodies without this core fucosylation can bind to FcgRIIIa with higher affinity, resulting in higher ADCC and efficacy.
The cell culture step is central to having the desired glycan profiles on recombinant proteins. Commonly, immature N-glycans, like high-mannose N-glycans, lack terminal sialyation and galactosylation and are more easily cleared in vivo, resulting in lower antibody efficacy. High-mannose N-glycans result from suboptimal CHO or HEK293 cell culture parameters.
“Although MS analysis is still the gold standard for characterization, glycan profiling can be infeasible,” Person warns. “We produce over 6,000 research-grade, non-antibody protein products and do not have the ability to run them all through MS due to the low throughput and high cost.”
Recently developed benchtop techniques rely on Bio-Techne’s catalog of glycobiology enzymes and substrates to footprint both N– and O-linked glycans. “We can use neuraminidase to remove sialic acid or FUCA1 to remove fucose followed by fucosyltransferases and sialyltransferases to add fluorescently labeled sugars to glycans,” Person relates. “Labeled recombinant proteins can then be run on SDS-PAGE gels, and differences in sialylation, galactosylation, or fucosylation can be easily visualized to compare glycan footprints.”
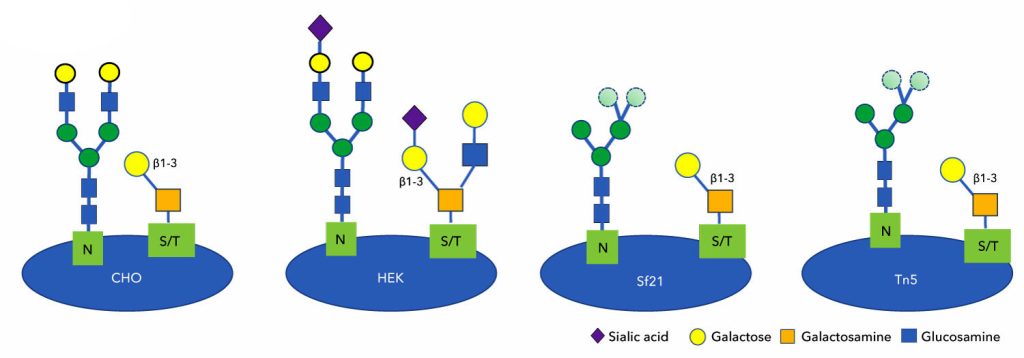
The approach was used to investigate differences in glycosylation patterns of SARS-CoV-2 spike proteins made in insect, CHO, and HEK293 production hosts.
The original SARS-CoV-2 canonical spike protein comprising the receptor binding domain (RBD) that was produced in insect hosts (Sf21 or Tn5) possessed generally high-mannose, immature N-glycans with more immature core-1 O-glycans. CHO host-produced RBD proteins showed N-glycans that were terminally galactosylated without terminal sialylation with core-1 O-glycans. The most mature RBD glycan profiles were seen on the HEK293 host-produced proteins that were both terminal sialylated on N-glycans and also displayed core-2 O-glycans.
“The finding that the original SARS-CoV-2 spike protein contains 22 N-glycosylation sites emphasizes glycobiology’s complexity,” Person declares.
Controlling microheterogeneity
“The importance of PTMs, particularly glycosylation, has been established in biopharmaceutical manufacturing,” says Nan Lin, PhD, director, New Technologies and Service Applications, Cytiva. Variants of glycosylation, together with variants in charge and size, are among the key aspects of recombinant protein microheterogeneity.
Controlling microheterogeneity to advance PTM control has become essential in implementing quality by design (QbD) and in meeting biosimilarity criteria. In recent years, industrial and academic researchers have made significant progress in controlling and modulating glycosylation via media and process optimization. “Glycosylation modulation,” Lin remarks, “has gained more focus not only in fed-batch but also in perfusion processes in recent years.”
Current methods are mainly based on rather lengthy workflows—collecting samples, purification, and analysis. High-throughput, low-sample-volume, and low-protein-quantity PTM analytical methods remain the challenge.
“High-throughput analytical methods, coupled with high-throughput cell culture platforms such as microbioreactors would enable assessment of PTMs in very early steps in cell line and process development,” Lin suggests. “Online/at-line analyses in an integrated, fully automated system has become increasingly discussed and developed for online PTM data monitoring.”
Numerous chip- and kit-based rapid glycosylation analysis methods are available and make good tools for bioprocess development, especially where analytical capabilities are not readily available.
“Several research groups have proposed to characterize global cellular PTM characterization, including glycosylation, phosphorylation, and ubiquitination, as part of understanding and controlling cell physiology,” Lin notes. “Such groundbreaking work would provide effective cell engineering targets and process monitoring endpoints in the future.”
Emerging biopharmaceutical molecules, including Fc-fusion and bispecific proteins, have gained presence in many pharmaceutical companies’ development pipelines. Many new molecules have unique structures and PTMs required by mechanisms of action.
“To be able to control PTMs of the new molecules early in development will be critical for future QbD processes,” Lin stresses. “Novel host cell lines for producing therapeutic proteins are on the horizon. Human origin or glycoengineered CHO host cell lines may be able to produce more human-like glycosylation patterns, and to achieve better glycosylation control.”
Exploring new ways to study low-abundance PTMs
“In vertebrates, the investigative focus has primarily been on serine, threonine, and tyrosine phosphorylation, but mounting evidence suggests that phosphorylation of other, ‘noncanonical’ amino acids also regulates critical aspects of cell biology,” reports a scientific team led by Claire E. Eyers, PhD, professor of biological mass spectrometry at the University of Liverpool.
“Eyers’ use of strong anion exchange-mediated phosphoproteomics to detect noncanonical phosphorylation indicated that the number of unique noncanonical phosphosites is approximately one-third of the number of observed canonical phosphosites,” comments Cristina Martin-Granados, PhD, research area scientific lead, Cell Signaling, Abcam.5 Although the numbers presented by Eyers and colleagues need to be firmed up, they are already encouraging researchers to consider broadening their views of the phosphorylation landscape.
According to Martin-Granados, low-abundance PTMs can make detection by antibody-based approaches difficult. Enrichment of a specific PTM by immunoprecipitation or by ion exchange, immobilized metal ion affinity, or immunoaffinity chromatography can help overcome low-stoichiometry challenges.
She emphasizes that dynamic and frequently transient PTMs require stringent positive and negative experimental controls if results are to be correctly interpreted. Since many PTMs are the aftermath of enzymatic reactions, uncontrolled enzymatic activity in sample processing can affect them.
“Antibodies are essential tools for detection and enrichment,” Martin-Granados remarks. “The challenge in developing highly specific antibodies with exquisite binding affinity is the small size of the PTM chemical moieties, similarities in some chemical structures, and poor antigenicity. Polyclonal antibodies present serious drawbacks in delivering reproducible and reliable data. To avoid these drawbacks, it may be necessary to switch to highly functional, reliable, and renewable recombinant monoclonals.”
Computational methods for predicting PTMs are attracting considerable attention. For example, AlphaFold, an artificial intelligence platform, made the news when it demonstrated its prowess in predicting unsolved protein structures from amino acid sequences. However, as Martin-Granados explains, AlphaFold2 does not yet consider the impact of PTMs on protein structure. Fortunately, databases on protein PTMs and predictive computational tools are available.
Proximity ligation assay (PLA), an antibody-dependent, fast, and highly sensitive immunoassay technology, can be used to detect PTMs, allowing for identification of several PTMs in a specific target.6,7 PLA technology can be used in combination with immunocytochemistry/immunofluorescence analysis and fluorescence immunohistochemistry analysis to study protein localizations and validate potential biomarkers for clinical diagnostic testing.
Genetic code expansion enables site-specific placement of functionally masked unnatural amino acids into proteins. These unnatural amino acids may remain inert until they are activated by a signal of some kind, that is, by a particular kind of illumination or by a small molecule. This provides rapid and temporal external control over addition and removal of PTMs, eliminating compensatory mechanisms that arise by slow conventional approaches, and it is expected to significantly increase the understanding of transient, dynamic PTM events.8
“Sensitivity and the dynamic range for detection of low-abundance PTMs need improvement as well as accurate, quantitative methods for studying dynamics and for analysis of less-well-known or newly identified PTMs,” Martin-Granados adds. “The relevance of multisite, cooperative PTMs highlights the need to develop strategies for full-spectrum identification of PTMs.”
Meeting characterization challenges
Previously, PTMs were thought of in binary terms—as either activating or inactivating, based on the particular modification and pathway. “Now, we understand to a greater degree how subtle changes in PTMs can modulate activity and signals,” says Carl Ascoli, PhD, chief science officer, Rockland Immunochemicals.
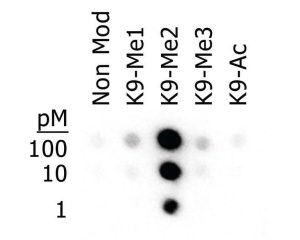
Although PTMs of proteins increase the functional diversity of the proteome to adapt to rapid changes in the environment, this diversity places strain on the availability of detection and characterization antibodies to certain modifications.
“Consider two characterization challenges: those involving ubiquitination, and those involving epigenetics,” Ascoli suggests. “Ubiquitination is a complex, multistep, reversible process that is highly regulated by the sequential action of very specific enzymes that add or remove ubiquitin. These enzymes may add monoubiquitin, polyubiquitin, and branched-chain ubiquitin at one or more targets sites on a given protein. This complexity makes understanding the machinery of ubiquitin in the laboratory a difficult process.”
“Epigenetic modifications, such as PTMs on histones, can be numerous, diverse, subtle, and transient, and they can work in combination,” Ascoli continues. Different cellular processes are affected depending on whether a particular amino acid is modified by a single, double, or triple methylation event, or whether it is unmethylated. Subtle variations in PTMs can result from changes due to environment, diet, disease, stress, or drug therapy.
Immunoassays using specific antibodies to PTMs generated in context with the surrounding amino acids on an enzyme are valuable tools for detection and characterization whether experiments are conducted in vitro or with biological systems involving immunofluorescence microscopy of organoids or whole animal in vivo imaging.
According to Ascoli, antibodies to AKT pS473, MEK pT386, or STAT3 pY705 have been invaluable in elucidating regulatory pathways and exploring the efficacy of drugs in both benchtop and biological experiments. He adds, however, that PTMs are more diverse and abundant than well-characterized antibodies.
Understanding the function of PTMs requires a more universal detection strategy. For instance, it may be necessary to rely on physicochemical detection methods such as MS analysis. Methods that look at entire proteins may be especially useful. Variations that increase accuracy and add context to the MS data include MALDI-MS, ESI-MS, and tandem MS.
“Detection or analysis methods that are based on protein binding directly or indirectly to DNA or RNA are of interest and will continue to evolve,” Ascoli predicts. “While assays like CUT&RUN, CUT&TAG, and more recent variations called TIP-seq and MULTI-CUT&TAG are themselves dependent on antibodies that bind to the protein that binds nucleic acids, the information that these assays provide is much richer in context.”
References
1. Aebersold R, Agar JN, Amster IJ, et al. How many human proteoforms are there? Nat. Chem. Biol. 2018; 14: 206–214. DOI: 10.1038/nchembio.2576.
2. Ramazi S, Zahiri J. Post-translational modifications in proteins: resources, tools and prediction methods. Database (Oxford) 2021; 2021: baab012. DOI: 10.1093/database/baab012.
3. Levene P, Alsberg C. The cleavage products of vitellin. J. Biol. Chem. 1906; 2: 127–133.
4. Kafader JO, Beu SC, Early BP, et al. STORI Plots Enable Accurate Tracking of Individual Ion Signals. J. Am. Soc. Mass Spectrom. 2019; 30: 2200–2203. DOI: 10.1007/s13361-019-02309-0.
5. Hardman G, Perkins S, Brownridge PJ, et al. Strong anion exchange-mediated phosphoproteomics reveals extensive human non-canonical phosphorylation. EMBO J. 2019; 38: e100847. DOI: 10.15252/embj.2018100847.
6. Tong QH, Tao T, Xie LQ, Lu HJ. ELISA–PLA: A novel hybrid platform for the rapid, highly sensitive and specific quantification of proteins and post-translational modifications. Biosens. Bioelectron. 2016; 80: 385–391. DOI: 10.1016/j.bios.2016.02.006.
7. de Oliviera FMS, Mereiter S, Lönn P, et al. Detection of post-translational modifications using solid-phase proximity ligation assay. N. Biotechnol. 2018; 45: 51–59. DOI: 10.1016/j.nbt.2017.10.005.
8. Zhou W, Deiters A. Chemogenetic and optogenetic control of post-translational modifications through genetic code expansion. Curr. Opin. Chem. Biol. 2021; 63: 123–131. DOI: 10.1016/j.cbpa.2021.02.016.