
February 15, 2013 (Vol. 33, No. 4)
Master Mix Method for DNA Fragments
Site-directed mutagenesis (SDM) is a commonly used technique for introducing mutations into a gene of interest. Existing techniques for SDM, such as whole plasmid SDM, while effective, are time consuming and prone to off-target mutation incorporation. Further, verification of mutation incorporation can be difficult when the insertion site sequence lacks convenient restriction sites for analysis. This can be a serious impediment to the planning and execution of SDM experiments.
Gibson Assembly™, developed by Daniel Gibson and his colleagues at the J. Craig Venter Institute, is a rapid and reliable method for the assembly of DNA fragments in a single-tube, isothermal reaction without reliance upon the presence of restriction sites within the target sequence. The technique, which involves the design of complimentary flanking primers to align fragments, can be readily adapted for SDM applications. In addition, it is unnecessary to use phosphorylated primers for Gibson Assembly, reducing both cost and time.
In one step, Gibson Assembly can assemble two or more PCR products with overlapping ends together or into a pre-cut vector. An exonuclease creates single-stranded 3´ overhangs that promote annealing of complementary fragments at the overlap region. A polymerase then fills in the gaps, which are sealed by the DNA ligase. By introducing multiple complementary mutations in the primers at the overlap region, the Gibson Assembly Master Mix forms a single, covalently bonded DNA molecule that contains the desired mutations and can be directly transformed into competent cells and screened or sequenced.
Here we describe the use of New England Biolabs’ Gibson Assembly Master Mix in two different mutagenesis experiments: first, multiple mutations of the lacZ gene (Figure 1) and second, the mutation of 5 of the 6 nucleotides at position 174–179 of eGFP from CTGACC to TTCTAT in order to change the amino acid sequence from LeuThr to PheTyr (Figure 2).
Single Bases at Multiple Sites
In this experiment, multiple primers were designed to incorporate three mutations within the gene. First, fragments with designed mutations in the primers were PCR amplified using the following primer pairs: lacZ-F1/R1, lacZ-F2/R2, lacZ-F3/R3, lacZ-F4/R4, and T7-For/T7-Rev. The resulting amplicons contained 18–20 bp overlaps and the desired mutations (Figure 1). Following PCR, 1 μL of DpnI was added to each tube and incubated at 37°C for an additional 30 minutes. After DpnI treatment, all products were cleaned up using Qiagen QIAquick™ PCR purification columns.
The concentration of the fragments was determined by Nanodrop™ instrument or estimated by agarose gel electrophoresis and then an equimolar amount of DNA fragments were added together up to 10 μL. NEB 2X Gibson Assembly Master Mix was then added and the reaction was incubated at 50°C in a thermocycler for 1 hour. 2 μL of the reaction was transformed into 50 μL of NEB 5-alpha Competent E. coli (High Efficiency), plated on LB-Amp plates and incubated overnight at 37°C.
Ten colonies were screened by sequencing. Eight contained the desired mutations.
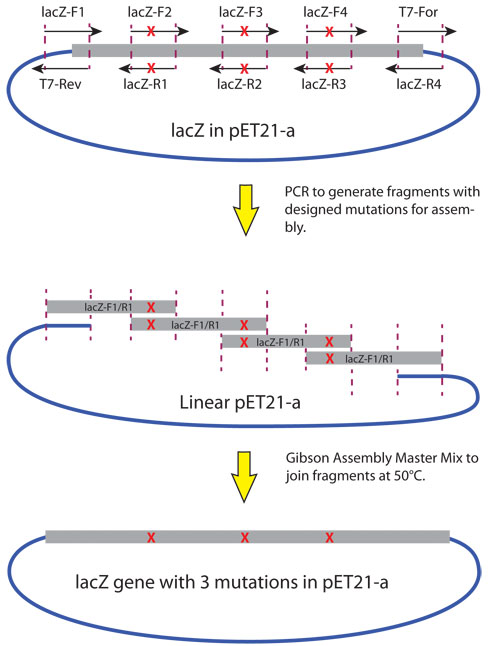
Figure 1. Site-directed mutagenesis of lacZ using Gibson Assembly. In this example, multiple mutations are introduced into the lacZ gene through overlapping primers followed by PCR. Gibson Assembly is then used to join the fragment with linearized vector.
Multiple Bases at a Single Site
For this experiment, two overlapping primer sets were designed to incorporate appropriate mutations to alter the two amino acid residues. After amplification, the two resulting amplicons (210 and 528 bp) will overlap by 24 bp and contain the desired mutations (Figure 2). Qiagen QIAquick PCR purification columns were used to clean up PCR products. The 5´ end of the first amplicon and the 3´ end of the second amplicon overlap with the vector sequence, which has been digested with NcoI and XhoI at 37°C for 2 hours. Restriction endonucleases can be inactivated by incubation at 65°C for 20 minutes. No gel purification is necessary, as the Gibson Assembly Master Mix will not religate the linearized vector.
Concentrations of PCR fragments were estimated by agarose gel electrophoresis. The Gibson Assembly reaction was set up with NEB 2X Gibson Assembly Master Mix, restriction enzyme digested vector, and two PCR fragments. The reaction was incubated at 50°C in a thermocycler for 1 hour. 2 μL of the reaction was transformed into 50 μL of T7 Express Iq Competent E. coli (High Efficiency, NEB# C3016), plated on LB-Amp plates and incubated overnight at 37°C.
Ten colonies were screened by sequencing. Five contained the desired sequence changes.
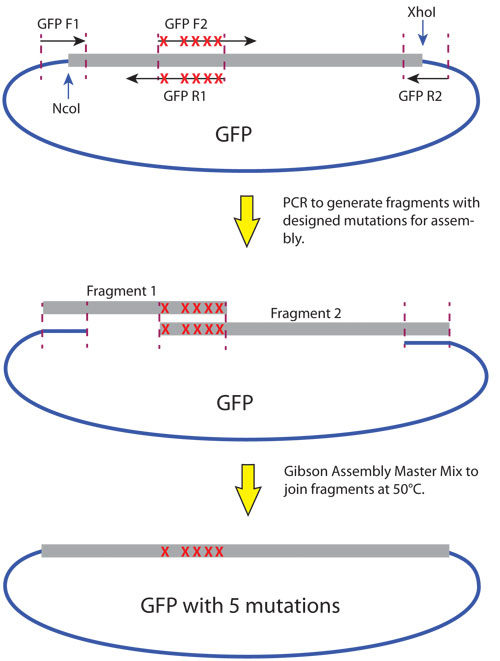
Figure 2. Site-directed mutagenesis of eGFP using Gibson Assembly. In this example, 5 nucleotides were changed by introducing the mutations into the overlapping primer, followed by PCR. Gibson Assembly is then used to join the fragments with enzyme-digested vector.
Summary
These results demonstrate the versatility of the Gibson Assembly Master Mix in both single and multiple site-directed mutagenesis.
In the first example, three mutations were introduced into the lacZ gene simultaneously. Four overlapping PCR amplicons were assembled with a linearized vector in one step. Resulting colonies were screened by sequencing. 80% of the colonies contained the desired mutations. This represents a substantial improvement upon earlier methods of multisite mutagenesis. Whereas previously, one may have had to create mutations sequentially, a significant increase in the length of the experiment, SDM using Gibson Assembly can be done in one step, and in much less time.
In the second example, the Gibson Assembly Master Mix was used to successfully incorporate five single base changes within a six base sequence of the GFP gene. Two PCR amplicons, overlapping at the location of the altered bases, were assembled with a restriction enzyme-cleaved vector in one step. The resulting colonies were screened by sequence analysis, which demonstrated that 50% of the colonies contained the desired sequence change.
In both cases, the Gibson Assembly Master Mix represents a substantial improvement over traditional methods, specifically in time savings, ease-of-use, and cost.
Ezra Schildkraut, Ph.D., and Peichung Hsieh, Ph.D ([email protected]), are both applications and product development scientists at New England Biolabs.

