
January 15, 2010 (Vol. 30, No. 2)
Shotgun Mutagenesis Mapping Provides Alternative to Site-Directed Techniques
Characterizing the binding sites of monoclonal antibodies (mAbs) on target antigens, their epitopes, can aid in the discovery and development of new therapeutics, diagnostics, and vaccines. For example, epitope mapping can aid in the selection of optimized therapeutic mAbs, elucidate cancer-specific epitope markers, and define the protective (and in some cases pathogenic) effects of vaccines. Epitope information can also help elucidate mAb mechanisms of action and strengthen intellectual property claims.
However, epitope mapping can be challenging for targets with complex structures. Such targets include transmembrane proteins (e.g., GPCRs, ion channels, and viral membrane proteins), eukaryotic proteins that are post-translationally modified, and multisubunit oligomers. This tutorial addresses the challenges for experimental techniques that enable rapid analysis of mAb epitopes and discusses solutions to the epitope-mapping bottleneck for complex targets.
Technical approaches for obtaining relevant mAb epitopes face several challenges. First, an epitope-mapping technique should be versatile enough to map both linear and conformational epitopes. Linear epitopes are formed by a continuous sequence of amino acids in a protein, while conformational epitopes are composed of amino acids that are discontinuous in the primary sequence but are brought together upon three-dimensional protein folding. Since many therapeutically important mAbs target conformational epitopes formed only in the native structure of the protein, the ability to map conformational epitopes is crucial.
Second, an epitope-mapping technique should map epitopes at high resolution so that individual amino acid contact points can be identified. Finally, epitope mapping must be applicable to high-value conformationally complex pharmacological targets, including membrane proteins, proteins that are post-translationally modified, and oligomers.
The gold-standard approach to epitope mapping is x-ray co-crystallography of a mAb together with its target protein. This technique allows direct visualization of the interaction between the antigen and mAb, and identifies both strong and weak contributions to the interaction.
Co-crystallography, however, is technically challenging to perform, and the high expense and time commitment involved precludes its application to many significant targets. Importantly, crystallography, as well as many other in vitro mapping approaches, relies on the availability of purified proteins (usually from prokaryotic sources), and is thus difficult to routinely apply to complex eukaryotic proteins such as multiple-spanning membrane proteins.
Commercial services for epitope mapping often employ peptide scanning. Here libraries of short peptide sequences from overlapping segments of the target protein are tested for their ability to bind antibodies of interest. The strategy is rapid, high-throughput, and relatively inexpensive to perform. However, since peptides are usually unable to mimic complex protein conformations, this strategy is applicable to mapping only linear not conformational epitopes.
Hydrogen-deuterium (H/D) exchange involves mapping epitope regions that are shielded by bound mAbs from the solution exchange of hydrogen and deuterium atoms. The protein is then digested into fragments that identify the protein region encompassing the epitope. Like crystallography, H/D exchange is compatible with mapping both linear and conformational epitopes but requires a purified source of protein. The technique is able to map mAb epitopes with moderate resolution by identifying protein fragments encompassing the epitope but is unable to identify specific amino acid contact points with high resolution.
Yeast display can be used to map the epitopes on some types of conformationally complex proteins. In this technique, libraries of randomly mutated protein fragments are displayed on the surface of yeast cells. Epitopes are then mapped by screening mAbs against these fragments using selective binding assays. The technique is relatively rapid, inexpensive, and compatible for mapping both linear and conformational epitopes. Whole proteins can not always be expressed, however, so some epitopes can be missed. In addition, proteins with complex structures, including multiple-spanning membrane proteins and oligomers, cannot be mapped using this strategy.
For many conformationally complex targets, the only epitope-mapping technique available is conventional site-directed mutagenesis and functional analysis in mammalian cell culture. This involves targeted site-directed mutagenesis where critical amino acids are identified by systematically introducing substitutions along the protein sequence and then determining the effects of those changes on mAb binding. The underlying limitation of this approach is that it is labor-intensive and slow, usually precluding the analysis of more than a handful of amino acids.
Shotgun Mutagenesis Mapping
To address the bottleneck in epitope mapping of complex proteins, a novel strategy, Shotgun Mutagenesis Mapping, was developed by Integral Molecular. This high-throughput mutation analysis strategy provides a complete, residue-by-residue evaluation of a target protein (Figure 1). Epitope mapping using Shotgun Mutagenesis begins with the creation of a plasmid-mutation library for the target gene, with each clone in the library bearing a unique amino acid mutation.
The clones that constitute the mutation library are individually arranged in microplates, expressed within living mammalian cells, and tested for immunoreactivity with mAbs of interest. Amino acids critical for mAb epitopes are identified by a loss of reactivity and are then mapped onto a protein structure to visualize epitopes. By automating the analysis, new epitope maps can be derived within days to weeks. Because it uses the native structure of proteins within mammalian cells, the technique allows both linear and conformational epitope structures to be mapped on complex proteins.
The utility of Shotgun Mutagenesis Mapping has recently been demonstrated using a well-characterized membrane protein target, CCR5. CCR5 is a eukaryotic GPCR widely studied for its role as a co-receptor for HIV entry. The relatively large number of commercial mAbs raised against this receptor, and a substantial body of published data describing its epitope reactivity, provided the ability to verify that Shotgun Mutagenesis Mapping results are accurate and comprehensive.
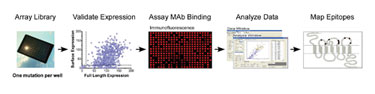
Figure 1. Each well of a Shotgun Mutagenesis mutation array contains a plasmid clone with a defined amino acid change.
A panel of primarily conformational mAbs was mapped using the technique. Shotgun Mutagenesis identified all of the 13 critical residues in these epitopes that were identified by others, as well as 11 novel critical residues that were not previously identified (Figure 2), thus validating the approach. Interestingly, because the arrays involved in Shotgun Mutagenesis are assayed using living mammalian cells, these same arrays have also been used to map other functional areas of the GPCR, including ligand activation regions, HIV co-receptor usage, and small molecule binding sites.
The ability to map mAb epitopes efficiently, accurately, and on structurally complex targets is a major advancement for the discovery and development of antibody therapeutics and recombinant vaccine strategies. New technologies for epitope mapping such as Shotgun Mutagenesis allow this critical information to be readily obtained for important therapeutic targets including structurally complex proteins.
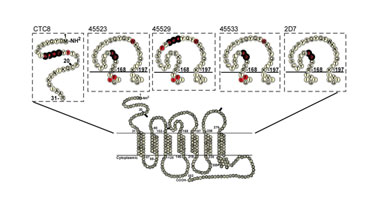
Figure 2. Epitopes of five CCR5 mAbs were mapped using Shotgun Mutagenesis.
Soma S.R. Banik, Ph.D., is a research scientist and communications specialist, and Benjamin J. Doranz, Ph.D. ([email protected]), is president and CSO of Integral Molecular. Web: www.integralmolecular.com.

