
September 1, 2011 (Vol. 31, No. 15)
Technology Owes Rapid Growth to Potential in Biomedical Research and Clinical Medicine
In recent years, next-generation sequencing approaches have generated unprecedented advances. With over 2,000 genes linked to at least one disease-relevant mutation, DNA sequencing has assumed increasingly important clinical relevance.
Nevertheless, multiple considerations still make it impractical to routinely sequence large numbers of eukaryotic genomes. Enrichment for specific regions of interest often becomes necessary, and targeted resequencing, which examines a limited number of genes within large populations, is emerging as a key approach, instrumental in our ability to unveil clinically and biologically relevant sequence variants.
Hanlee P. Ji, Ph.D., assistant professor in the department of medicine at Stanford University, and colleagues have developed several different approaches that allow targeted resequencing of genomic DNA and are useful for many applications such as the validation of mutations from cancer genomes, a topic that is still filled with challenges, partly due to the genetic heterogeneity found in clinical samples.
“This is one application for which targeted resequencing will be necessary and is unlikely to go away anytime soon,” explains Dr. Ji.
Another application is in clinical diagnosis, when multiple mutations in several genes are often linked to the same malignant tumor, but interpreting the clinical relevance of individual mutations is often a difficult task.
“I think what is practically going to happen is that gene subsets that have immediate clinical relevance and are clinically actionable will be quickly disseminated as diagnostic tests, and we already have started seeing that.”
Dr. Ji and colleagues recently published a targeted resequencing strategy that uses in-solution 80-mer oligonucleotides for capture and allows the analysis of gene subsets from the human exome. “This first-generation targeting technology is public and openly available for users.”
The Human OligoExome application is available at oligoexome.stanford.edu. Users can download the capture oligonucleotides to create their own assays independently of working with commercial sources.
“I think this is the first time that something like this has become available, on such a scale, for targeted resequencing,” explains Dr. Ji. The authors demonstrated the general robustness of this particular technology and revealed that it can resequence, with high sensitivity, genomic regions up to 1 megabase in size, when as little as tens of nanograms of target DNA is available.
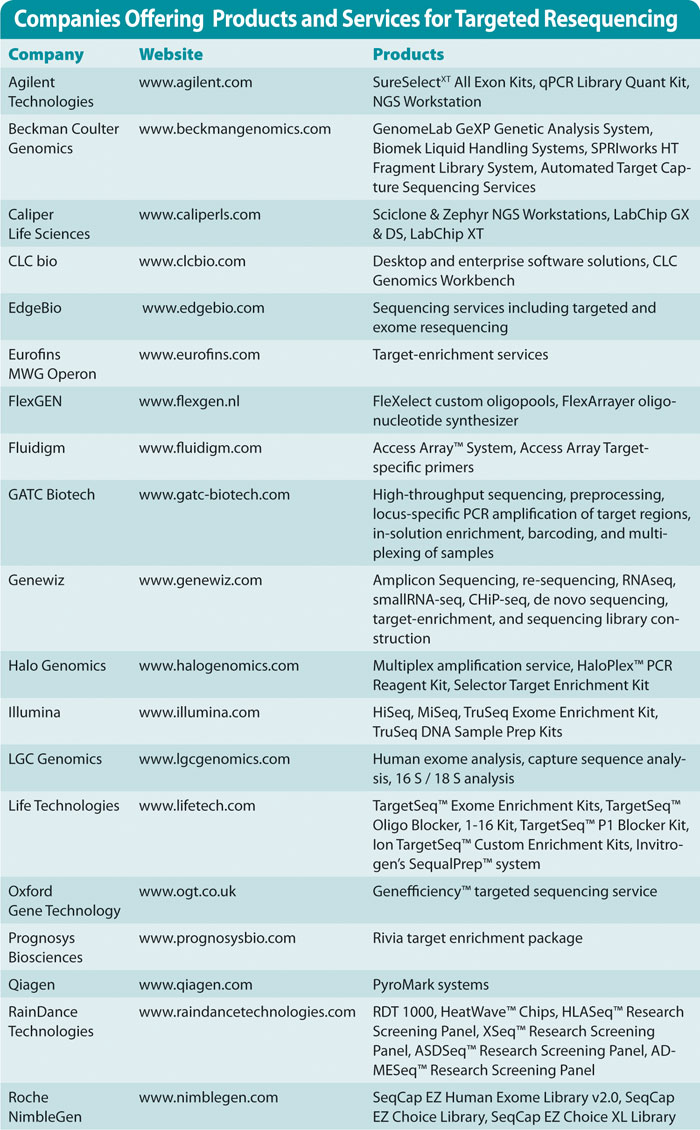
Companies offering products and services for targeted resequencing
New Approaches
While targeted resequencing provides a revolutionary approach to survey genetic variants, it also presents specific limitations for clinical diagnostics. “One challenge is that the existing targeted methods, which still require a large amount of sample and are expensive, are very cumbersome for molecular diagnostics,” says Patrice Milos, Ph.D., svp and CSO at Helicos BioSciences.
Dr. Milos and colleagues recently developed an approach that allows any gene to be captured and sequenced directly from genomic DNA, eliminating the need for DNA amplification and for other enzymatic steps prior to sequencing.
DNA isolated from a clinical sample of interest is initially sheared and then introduced into the custom flow cell for sequencing, and all these sample-prep steps can be performed within two hours. “In addition, what is being sequenced is the patient’s natural DNA, as opposed to DNA obtained by methodologies that involve amplification and other enzymatic steps.”
This method involves very small sample amounts, and Dr. Milos illustrated its accuracy and low cost in a recently published study that examined mutations in the BRCA1 gene. An important advantage that this approach presents over Sanger sequencing and enzymatic amplification techniques is the quantitative nature of the sequencing, which enables it to identify not only substitution mutations but also large insertions, deletions, and rearrangements.
Data Analysis
“The field of human genetics is highly dynamic. Every few years, there is a major technological change, and it is important to adapt to these new technologies and devise novel approaches to analyze the data that is coming out,” says Chun Li, Ph.D., an investigator at the Center for Human Genetics Research at Vanderbilt University.
While genome-wide association studies have seen unprecedented advances, these approaches still face several challenges. One of them is the difficulty inherent in sequencing the genome of every participating individual. This is particularly cumbersome in the case of rare disease alleles and uncommon genetic variants for which resequencing is currently the only feasible strategy to reliably identify them and interrogate their clinical relevance.
“Once we are able to perform sequencing at the highest resolution, one of the greatest challenges is how to analyze the data,” reveals Dr. Li, who, together with colleagues, developed SampleSeq, a probability-based algorithm that allows investigators to select and optimize samples for targeted resequencing.
The authors revealed that in groups of unrelated study participants, SampleSeq enriches the yield of rare or uncommon disease alleles and helps select participants with a balanced representation of all chromosomal regions when multiple regions of interest have to be sequenced. In addition, SampleSeq can estimate the sample size that is necessary to detect a rare allele and can thus guide resequencing efforts.
RNA-Seq
As traditional chain terminator or Sanger DNA sequencing shifts toward next-generation and next-next generation approaches, reads are becoming available at increased throughput and lower costs. Applying these methods to cDNA libraries has catalyzed the emergence of RNA-Seq, a new tool that enables an unprecedented level of visualization of the eukaryotic transcriptome.
RNA-Seq is informative about multiple levels of the genome transcription, including DNA sequence, gene expression, and mRNA splicing, which cannot be interrogated by using any other single approach.
“One of the key things about RNA-Seq is that the information it provides cannot be obtained from genomic DNA sequencing,” says Joshua Z. Levin, Ph.D., research scientist and group leader in the genome sequencing and analysis program at the Broad Institute.
One of the challenges associated with whole transcriptome sequencing is the different level at which each transcript is present. While good coverage can be obtained for highly expressed transcripts, this is more difficult in the case of low-level transcripts for which resequencing is needed to achieve sufficient coverage and to ensure that an observed change is relevant and not merely a sequencing error.
“We need to have enough coverage, and we often need to see specific regions sequenced 10 to 20 times at a given position to be confident that the sequence is there, particularly when a genetic variant could exist in a heterozygous form,” says Dr. Levin.
In an approach known as targeted RNA-Seq, Dr. Levin and colleagues combined next-generation sequencing with the hybridization capture of relevant transcriptome subsets. The authors revealed, in a study that used oligonucleotide probes specific for 467 cancer-related genes from a tumor cDNA library, that targeted RNA-Seq increased the coverage of low-abundance transcripts to levels that allowed sequence changes to be easily detected.
Targeted RNA-Seq provides information that cannot be obtained by any other single method. Learning about alternative splicing, identifying novel fusion transcripts, quantitating transcript levels, and providing insights into allele-specific expression are some of its greatest advantages. When two different alleles co-exist, DNA sequencing alone cannot determine which one is expressed and to what proportion, but this information may be very relevant clinically.
Anthropological Applications
An area that has been profoundly impacted by targeted resequencing is anthropology. Svante Pääbo, Ph.D., director in the department of genetics at the Max Planck Institute for Evolutionary Anthropology, and colleagues recently revealed, in studies that examined bones from the Vindija cave in Croatia, that Neanderthals, the closest evolutionary relatives of present-day humans, share more genetic variants with European and Asian populations than with individuals from sub-Saharan Africa.
One of the major challenges that investigators faced is the extensive contamination of the DNA of interest with microbial DNA. “The high proportion of microbial DNA made shotgun sequencing impractical,” says Dr. Pääbo. By using targeted resequencing on ~49,000-year-old Neanderthal bone remains obtained from the El Sidrón cave in Spain, Dr. Pääbo and colleagues were able to successfully recover more than one megabase of target DNA regions.
“The targeted approach and the capture approach are really important in ancient DNA research, not only because they allow the capture of a certain part of the Neanderthal genome but also because the Neanderthal DNA represents approximately ~0.2 percent of the total DNA we have,” explains Dr. Pääbo.
The investigators interrogated approximately 14,000 protein-coding positions and identified 88 amino acid substitutions that became fixed in the human population since divergence from Neanderthals, illustrating the strength of targeted resequencing in shedding light on human protein evolution and in obtaining information from heavily contaminated ancient DNA samples.
By relying on several experimental approaches, including array-based and solution-based capture, Dr. Pääbo and colleagues are working on a survey of all the single-copy parts of chromosome 21. “We are seriously working on scaling up these techniques to eventually try to capture the whole single-copy component of the Neanderthal genome.”
While PCR has long been able to explore specific chromosomal regions, targeted resequencing not only provides insight about the chromosome at a much larger scale but also comes with additional advantages. For example, PCR amplification, which uses two primers, is very sensitive to mutations that could exist in the target at the sites where the primers anneal. “When things are captured by hybridization, the whole target is being used. Substitutions in the target have, therefore, a much smaller impact, and cause fewer problems than old-fashioned PCR that we used to perform,” explains Dr. Pääbo.
Targeted resequencing provides a more unbiased perspective of the genomic regions of interest and is not so sensitive to polymorphisms or other genetic variants.
There is no question that targeted resequencing is having an impact. It is important to appreciate the advantages that each individual technique offers, the multiple layers of information that can be interrogated by combining different approaches, and the fascinating biological questions that find answers and fuel even more questions, illustrating the dynamic nature of scientific inquiry.

Last year Svante Pääbo and his team from the Max Planck Institute for Evolutionary Anthropology reported the sequencing of an initial draft of the Neanderthal genome. The scientists generated the sequence from several fossils from Croatia, Germany, Spain, and Russia using high-throughput technologies.[Volker Steger/Photo Researchers]

