
January 15, 2010 (Vol. 30, No. 2)
In Silico Tools Can Augment Data-Gathering Pharmacokinetic and Toxicology Experiments
Researchers and patients have a common desire for safe and efficacious medicines. As a result, all of the tools that aid in the process of creating new drug products are highly valued. Software tools won’t replace data-gathering pharmacokinetic and toxicology experiments; however, researchers can leverage the data mining and computer modeling capabilities using already amassed information to make predictions to facilitate drug discovery and development.
Understanding and modeling the basic absorption, distribution, metabolism, and elimination (ADME) processes for organisms exposed to drugs contributes to the safety assessment of a drug product, which requires not only the evaluation of a drug substance, but also safety profiles of its metabolites.
The ability to predict the potential risk associated with metabolite vulnerabilities can be used to help redesign and select more promising candidates for further and more rigorous testing throughout the phases of discovery and development. Obviously, simple and confident estimation of candidate risk is desired.
In vitro studies, however, still involve extrapolations, and conducting all experimental tests for candidates would require prohibitive investments of animals, time, and money. Overall costs should be alleviated by early rejection of compounds with problematic metabolites and reallocation of resources toward more promising candidates. These factors increasingly emphasize the value of reliable in silico property prediction as part of the process.
Each organization has its own preferred and dynamic battery of experiments and computational assessments for new chemical entities and derived metabolites, including objectives to:
- Predict basic physicochemical properties (logP/logD, pKa) that determine the distribution of metabolites in an organism and their clearance;
- Predict the likelihood that a metabolite will be further metabolized by cytochrome P450 enzymes;
- Predict inhibition of cytochrome P450 isoforms, and thereby, possible interactions of metabolites with concomitant drugs;
- Predict hERG inhibition, genotoxicity, and other toxicity endpoints.
Despite the complexity of biological systems in organisms, software enabling the prediction of ADME properties from molecular structure is available. While a variety of approaches to individual predictions have been expounded, it is possible to find an extensive set of prediction capabilities, including those addressing the objectives mentioned in this article. The use of logP, logD, solubility, and pKa for predicting ADME properties such as permeability, oral bioavailability, and distribution are well documented and will not be discussed further.
Hepatic metabolism by cytochrome P450 enzymes is the major clearance route for xenobiotics. These biotransformations can lead to either an increase or decrease in toxicity. Using in silico tools such as the cytochrome P450 predictive modules in the ACD/ADME Suite from Advanced Chemistry Development (ACD/Labs), researchers can identify whether compounds will be substrates and/or inhibitors of the five major drug metabolizing enzymes—CYP3A4, CYP2D6, CYP2C9, CYP2C19, and CYP1A2.
Furthermore, the regioselectivity module predicts the sites most likely susceptible to metabolism in human liver microsomes and proposes possible biotransformation reactions. These predictors also offer the ability to add experimental data to extend chemical space coverage and improve prediction accuracy.
Drug-Drug Interactions
Awareness of drug-drug interactions can come from in silico tools by simple prediction of inhibitors and substrates of P450 enzymes, although mechanisms of interaction may be more complex.
Plavix (Clopidogrel bisulfate), a prodrug indicated for cardiovascular atherosclerotic disease, is metabolized in human organisms to produce an active metabolite that inhibits platelet aggregation. The active metabolite of Plavix is formed after the cytochrome P450-mediated opening of the thiophene ring, and ACD/ADME Suite predictions of metabolism regioselectivity are consistent with these experimental data.
In November 2009, the FDA recommended avoiding co-administration of Plavix and CYP2C19 inhibitor Omeprazole because the efficacy of Plavix is significantly reduced. After analysis of available experimental data, another compound, antiallergy drug Azelastine, was found to be a moderate CYP2C19 inhibitor (Ki=29 µM). The possibility of clinically significant drug-drug interactions is low for inhibitors of this potency.
The main metabolite of Azelastine, N-desmethylazelastine, inhibits CYP2C19 more effectively (Ki=7.3 µM). There is borderline potency for drug-drug interaction between the compound and CYP2C19 substrates such as Plavix. Therefore, the metabolite of Azelastine deserves attention. This entire situation is well reflected by ACD/ADME Suite predictions.
Problematic metabolites are disquieting for even commercially available, simple, well-known molecules such as acetaminophen. This drug has a generally safe metabolite profile, except for a minor metabolite accounting for Figure 1).
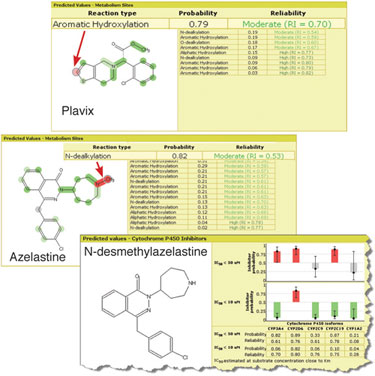
Figure 1. Results from ACD/ADME Suite P450 Inhibition and Regiosensitivity modules predicted sites of metabolism for Plavix and Azelastine.
Terfenadine, an antihistamine drug widely prescribed in the 1990s, was found to exhibit cardiotoxicity due to hERG channel inhibition only after it came to market. Terfenadine undergoes metabolism to Fexofenadine, which is also a potent histamine antagonist but does not inhibit hERG. This metabolite replaced the parent drug after it was famously withdrawn from the market in 1998.
While the hERG-channel inhibition assay has become one of the standard early tests in drug discovery, the backlog of early screening can be alleviated using in silico tools. Running these two compounds through the ACD/Tox Suite hERG Inhibitors module, we see that Terfenadine would be expected to be a hERG inhibitor (predicted hERG inhibition probability 0.98) while Fexofenadine would not (predicted probability 0.16), both predictions being highly reliable (Figure 2).
Genotoxicity of chemicals also often involves metabolic activation. For example, polycyclic aromatic hydrocarbons are only weak mutagens, but their metabolites, mostly epoxides, are strong mutagens. Therefore, genetic toxicity experiments, like the Ames test, examine compounds with and without metabolic activation. Given this fact, ACD/Tox Suite predicts genotoxicity while taking metabolic activation into account.
When using in silico tools, it is important to have realistic expectations of results. During the evaluation, one should keep in mind that a QSAR model is only as good as the quality of the associated data and is applicable only for the chemical space covered by the training set. For some desired endpoints there may be little available experimental data upon which to build a viable model.
When enough data is available, its quality may be insufficient to build a reliable model. The best scenario for building a predictive model is to use a global dataset, with local models “sitting on top” applying corrections for a series of structurally similar compounds. Ideally, the user could add in-house data to improve prediction accuracy. The trainable model feature of many ACD/ADME Suite and ACD/Tox Suite modules allows such customization of models.
Reliability of predictive models will continue to be an area for development and improvement. With drug discovery and development organizations facing increasing pressure to deliver investigational new drugs with favorable safety and toxicity metabolite profiles, we anticipate greater application of in silico tools. They can support experimental efforts by predicting superior compounds for synthesis and reduce the burden on initial high-throughput screens.
These and other examples suggest that early prediction of possible metabolites and their properties may alert researchers to safety risks not directly associated with the drug substance itself but with products of its biotransformation. To avoid failures, especially in late development or after-market, the key would be to use all the tools at our disposal for metabolite safety research, including predictive software.
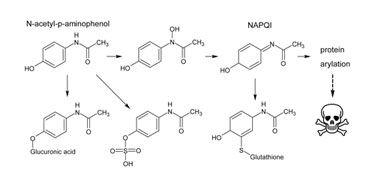
Figure 2. The main pathways of metabolism for acetaminophen are shown, including production of the minor toxic metabolite NAPQI.
Graham A. McGibbon, Ph.D. ([email protected]), is the mass spectrometry product manager for ACD/Labs, Pranas Japertas, Ph.D., is director of product development, ADME, physchem & tox, and Sanjivanjit K. Bhal, Ph.D., is a technical marketing specialist. Web: www.acdlabs.com.

