
February 15, 2012 (Vol. 32, No. 4)
Surface plasmon resonance (SPR) is not the new kid on the block. It’s been an industry workhorse technology for more than 20 years. What is new is coupling this premier technology with thermodynamic methodologies or molecular dynamics simulations. When used together, these provide a powerful means to generate a greater depth of information than either can alone. Further, the ability to sensitively gauge kinetics and equilibrium information positions use of SPR as an adjunct to high-throughput screening and affinity ranking during lead optimization.
The recent “Developments in Protein Interaction Analysis” conference hosted by GE Healthcare highlighted several of these synergistic partnerships as well as new ways SPR is being employed in the modern-day drug discovery arena.
Typical SPR requires very little material and does not need an external probe for monitoring interactions. This label-free, real-time technology is initiated by immobilizing a component, such as protein or nucleic acid, onto a sensor chip. The other component(s) are then injected over the flow cell(s) in the desired buffer. SPR detection relies on a resonance interaction between the light beam and the thin metal film on the sensor chip.
Allostery is the scientifically intriguing process in which the binding of a ligand to a macromolecule alters ligand binding at distant sites. Named by Nobelist Jacques Monod in the early 1960s to describe the sigmoidal binding curve of oxygen to hemoglobin, its molecular mechanisms have been a holy grail of biochemistry ever since.
“Scientists have spent decades trying to understand protein allostery through ligand-binding kinetics, thermodynamics, and structures, but we still do not have a unified picture,” said Jannette Carey, Ph.D., professor of chemistry, Princeton University. Besides its academic interest, allostery is thought by some to offer a new approach to drug discovery.
Dr. Carey’s laboratory uses a combination of experimental and computational techniques to study allostery in several protein-ligand systems.
“We initially quantify a binding process using biochemical and biophysical approaches, including isothermal titration calorimetry (ITC) and SPR. ITC measures the heat flow associated with a molecular interaction, complementing SPR by providing thermodynamic information.
“Recently we added molecular dynamics simulations (MD) to our arsenal, in collaboration with Professor Rüdiger Ettrich of the Czech Academy of Sciences. MD applies Newton’s laws of motion to molecules, providing a temporal description of structural changes.
“I was stunned when we saw that MD was able to trace an allosteric response from specific atomic detail to global conformational change.”
Experimentalists, including Dr. Carey, initially were skeptical of MD. “MD can be used to make experimentally testable predictions, putting it firmly in the realm of the scientific method. Besides requiring huge computational power, MD interpretation is complex and subtle, and thus a job for experts. So don’t expect an app for that any time soon.”
Dr. Carey’s take-home message is that “quantitative studies of ligand binding are the first step in understanding allostery and the biological roles of molecular interactions. In every course I teach I try to work ligand-binding theory and practice into the curriculum.”

Technician using a surface plasmon resonance machine to study biomolecular interactions. SPR involves the excitation of surface plasmons by light. A typical use for this technology is the characterization of antibodies. [Andrew Brookes, National Physical Laboratory/Photo Researchers]
DNA Therapeutics
With the identification and sequencing of most of the human genome in hand (and some disease-causing microorganisms), new opportunities to devise DNA-specific therapeutics are rapidly evolving.
“Gene-specific therapeutics require developing highly specific DNA-targeting molecules that recognize binding sites that are 10–15 bases long,” said W. David Wilson, Ph.D., professor of biophysical chemistry, department of chemistry, Georgia State University.
“Most of these are designed to target the minor groove DNA. The minor groove of DNA, as opposed to the larger major groove, is a better drug target since small molecules can more easily span this region.”
Dr. Wilson is utilizing SPR and microcalorimetry methods to design and evaluate compounds. “Our goals are to understand the details of DNA molecular recognition. DNA does nothing unless it interacts with another molecule. Designing small molecules that target the minor groove has the potential to shut down entire genes of pathogens, such as malaria.”
Dr. Wilson and colleagues designed both symmetric compounds (with two linked amidine-benzimidazole-phenyl units to specifically target AT sequences) and linked hairpin type heterocyclic compounds.
“Both types have the potential for enhanced binding specificity by recognizing both strands of duplex DNA. We use biosensor-SPR and microcalorimetry to evaluate the interaction of compounds with different DNA sequences.”
Coupling SPR and microcalorimetry provides even more information about compounds. “Microcalorimetry offers a means for much more detail relating to the affinity, and the mechanism of the reaction, such as its entropy and how that drives the reaction. It also provides information as to how chemical variations might contribute.”
An advantage of SPR is that it allows monitoring binding reactions in real time. “We use a gold-plated chip that can be functionalized with carboxymethyl dextran. We link streptavidin to that and couple biotin to the DNA. The binding of DNA to the chip will produce a signal change over a range of compound concentrations, allowing us to determine standard kinetics and equilibrium constants.”
Dr. Wilson found some surprises. “We studied such issues as the levels of DNA binding affinity that can be obtained with our relatively simple compound set, how compounds folded after binding, and the differences between compounds that bind to two sites versus one, etc.
“We are excited by our findings. We saw that a compound can do one of three things. First, it can fold back on itself while in the minor groove, and second, two compounds can—as expected—bind to different sites.
“But to our surprise, a third finding was that two compounds can form stacked dimers in the groove. We still don’t completely understand all of the dynamics, and will need high-powered binding methods to sort this out. But we believe this is something we can accomplish. Ultimately we then will pursue animal studies.”
NCE Decisions
Utilizing SPR as a follow-up to a high-throughput screening campaign provides an easy and reliable means to identify false-positive hits such those showing “promiscuous” binding behavior as well as nonbinders, suggested Jörg Bomke, principal scientist, molecular interactions and biophysics, NCE lead discovery technologies, Merck Serono Research & Development.
He discussed how small molecule interaction studies with SPR are utilized for the new chemical entities (NCE) drug discovery process at his company.
“Quite often the kinetics can explain discrepancies between the biochemical and cellular activities of diverse chemical scaffolds. Further, using SPR for fragment screening not only delivers high quality (well-behaving) hits, but also determines their affinities up to the millimolar range.
“We are titrating our fragments from 2 millimolar downward to assess their properties at this concentration and to sort out the misbehaving compounds. I believe this is very important for obtaining good success rates in crystallization trials with millimolar and higher micromolar fragment hits since a co-crystal structure determination needs a high occupancy of the binding site with the fragment.
“Ideally, the fragment concentration should be five to ten times higher than its affinity to the target.”
Bomke also explained how generating a kinetic profile helps with decision making. “Starting after a high-throughput screening run with scaffolds that exhibit already slow kinetics makes life easier for the medicinal chemists. Our experience shows that in some cases you cannot implement such a profile to your hit series even after prolonged chemical synthesis and optimization processes.
“In this respect, not only long residence times on the target are of interest, but also slow association rate constants and deviations from simple 1:1 models. Both of the latter can provide much information about the nature and mechanism of the drug-target interaction.”
For example, the importance of obtaining kinetic profiles at later stages of NCE drug development is the ability to identify lead candidates having a high affinity, but low “on” rates as well as low exposure levels or a high clearance.
“These candidates might not reach sufficiently high plasma levels over a longer period of time to induce an effect on their target protein, and therefore fail to show in vivo efficacy in animals or humans.”
It is also important to work with full-length proteins when possible. “One key observation over the last few years is that studying just one protein construct may lead to incorrect conclusions.
“By working with different protein constructs, mutants, post-translationally modified protein, and isoforms as well as using additional assays such as competition setups or co-injections (i.e., with co-substrates), one should gain a deeper insight to help reduce the gap between the in vitro world and the in vivo target physiology.”
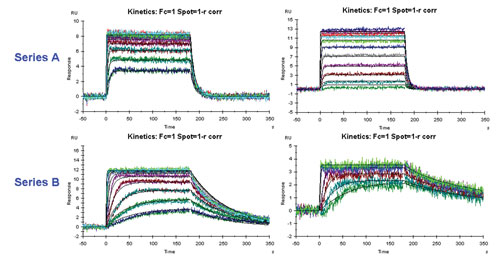
HTS hit series prioritization in drug discovery: NCE titrations on target protein surfaces: Hit series B shows a more desired kinetic profile; with its lower “off-rate”, this series might represent the better starting point (scaffold) for a drug development program. [Merck Serono Research & Development]
SPR and GPCRs
G protein-coupled receptors (GPCRs) are one of the most important classes of targets for drug discovery. They are linked to many disorders, yet the inability to apply structure-based drug design has hampered identification of novel therapeutic compounds, according to Andrei Zhukov, Ph.D., senior scientist, Heptares Therapeutics.
“Since GPCRs are very unstable when isolated from their native membrane environment, it is very difficult to crystallize them and reveal their structures by x-ray analysis. Without knowing the structure, rational drug design is very difficult. GPCRs also cannot be studied using biophysical methods such as SPR.”
Dr. Zhukov says that Heptares employs its StaR® (stabilized receptor) technology to introduce a small number of point mutations into GPCRs that increase their thermal stability in complex with a ligand to maintain the receptor in a specific physiological conformation. Amino acids are systematically mutated, and the small number of mutations having the highest stabilizing effect are selected and combined.
Stabilized receptors can be isolated from cells, purified without loss of ligand binding activity, and used for ligand interaction analysis by SPR.
“At the initial stage of drug design, SPR is used for hit identification by fragment screening. Later, during lead optimization, SPR is used for affinity ranking of the leads as well as to characterize the kinetics of drug-target interaction, which is known to have a strong impact on drug efficacy.”
Dr. Zhukov reported on a study in which several receptors were attached to Biacore (GE Healthcare) chips while a series of known binders and novel drug candidates were screened to obtain kinetic and affinity parameters.
“Another approach we use is Biophysical Mapping™ whereby binders from different chemical series are screened by SPR against binding-site mutants to define the role of active site residues in ligand binding and to refine binding models obtained using computational chemistry when the crystal structure is not yet available.
“Using this approach, important GPCRs, for the first time, have become tractable targets for drug discovery. For example, a highly selective agonist against the M1 receptor is advancing to the clinic as a potential treatment for Alzheimer disease and for improving cognition.
“Similarly, Heptares has identified a range of small molecule antagonist chemotypes to orexin 1/2 for treating sleep and potentially addiction disorders. Heptares also has ongoing lead discovery programs using the same approach to find GLP-1 agonists/allosteric modulators for diabetes and mGluR5 negative allosteric modulators for psychiatric disorders.”

