
June 1, 2015 (Vol. 35, No. 11)
Systems Biology Is No Longer a Fringe Discipline. It’s Getting to the Heart of Disease Processes
The “systems” in systems biology become apparent only when the whole is contemplated, not the parts—or even the sum of the parts. Mere accumulation cannot account for life’s complexity.
Rather, this complexity is emergent. It is more than the sum of the parts. It is the result of collective behaviors that cannot be reduced to the individual behaviors of simple components.
By modeling the interactions of biological components across multiple scales—from tiny feedback loops to far-flung networks—systems biology can depict complex and dynamic living systems more accurately than the traditional reductionist approach. Systems biology has been applied not only in bacteria and yeast, but also in plants and animals. In the past decade, systems biology has advanced so much that it has accomplished a shift: It is no longer a fringe discipline. It is now a mainstream science.
This transformation was much in evidence at a recent meeting, Systems Biology: Networks. Here, under the auspices of the Cold Spring Harbor Laboratory (CSHL), researchers discussed developments in this rapidly evolving field including novel approaches to understand gene regulatory networks, signal coordination, and the metabolome by integrating technology, computation, and critical human insights.
Gene Regulatory Networks
Plants have much to teach about biology and life. After all, Austrian biologist and monk Gregor Mendel provided the first clues to genetic inheritance and the nature of genes by meticulously crossbreeding pea plants. Further, American cytogeneticist Barbara McClintock received the Nobel Prize for work that revealed genetic transposition in maize.
Researchers are still utilizing plants to decipher gene regulatory networks. For example, Siobhan M. Brady, Ph.D., an associate professor at the University of California, Davis, utilizes the plant model Arabidopsis thaliana.
“A plant cell’s shape and function are largely determined by its cell walls. The primary wall surrounds the plasma membrane, and the secondary wall (such as those found in xylem) functions in water transport and protection against pathogens,” said Dr. Brady at the CSHL event. “It is also provides dietary fiber for humans, raw material for paper, and components for biofuel production.”
“Despite its importance, we know very little about precise regulatory mechanisms in time and space that give rise to the secondary cell wall’s three main components—cellulose, lignin, and hemicelluloses,” Dr. Brady admitted. Of the three main cell wall components, lignin is the most problematic. It impedes the extraction of cellulose and hemicelluloses for biofuel production.
The production of different cell wall components could be subject to manipulation. Already, ideas for how such manipulation could be accomplished are emerging from Dr. Brady’s group, which seeks to map the gene regulatory networks responsible for the biosynthesis of these components.
Such work, Dr. Brady indicated, requires a systems biology approach, the better to distill the large amount of data generated into organizing principles. She went on to describe her group’s particular systems biology approach: a combination of high-spatial-resolution gene expression data and a review of the literature on xylem cell specification.
“Instead of following single genes, we looked at the function of hundreds of transcription factors and enzymes in the regulatory network of xylem,” explained Dr. Brady. “We were able to derive hundreds of novel regulators and gain considerable insight into xylem developmental regulation.
“We found that under specific stress conditions, such as iron deprivation and salt stress, we could enhance production of individual components. This is important because it shows that regulation of these enzymes can be uncoupled and allows us to potentially manipulate the system to enhance production of biofuels.”
Dr. Brady is also examining this network in sorghum: “We’d like to understand how gene regulation networks are wired in the secondary walls of other species. Ultimately, we may be able to find existing plants that won’t require engineering to build better biofuels.”
Integrating Signaling Networks
Understanding the sophisticated communication networks in cells requires an in-depth knowledge of how cells integrate signals, noted Michael Springer, Ph.D., an assistant professor of systems biology at Harvard Medical School. Dr. Springer, who focuses on signaling network interactions, is studying the induction of galactose metabolic genes when budding yeast are grown in mixtures of glucose and galactose.
“This is a classical, well-studied system that one might think would yield no new insights, but this is precisely the perfect system for the type of research we do,” said Dr. Springer. “We use a combination of quantitative high-throughput assays with modern genetic approaches such as xQTL (a web platform that maps quantitative trait loci) and systematic deletion collections to analyze natural variants and mutant strains.
“Basically, we threw the kitchen sink at the problem: microscopy, flow cytometry, mass spectrometry—whatever we thought would help us answer our questions. Working on a system that is so well understood makes it possible for us to make sense of our results.”
According to Dr. Springer, there were many studies that looked at glucose repression of galactose genes, but none in quite the way they did: “Our strategies, combined with some simple models and quantitative approaches, are what led to our novel insights. Our key published result is that yeast respond to the ratio of glucose and galactose. This ratio sensing occurs upstream of the canonical signaling pathway, likely at the transporters, which are typically thought of as passive players in the response.”
But there are also some remaining challenges that Dr. Springer’s laboratory must still address: “I think this is a perfect system to start to hack away at the relationship between genotype and phenotype. There are many perspectives and angles to this that I think will keep us and others busy for a while. We are currently attacking the problem with a number of techniques. Over the next couple of years, I hope to be able to bridge these typically orthogonally used approaches. We hope this bridging will have a broad impact beyond just our knowledge of carbohydrate metabolism in yeast.”
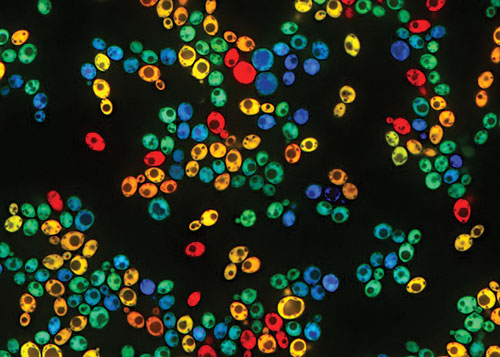
Scientists at Harvard Medical School are studying galactose metabolism by growing natural yeast isolates in mixtures of glucose and galactose. Strain BC187 has been engineered to constitutively express mTagBFP2 (blue cells), whereas strain YJM978 constitutively expresses mCherry (red cells). Both strains also express GAL1pr-YFP as a transcriptional reporter in the galactose-utilization (GAL) pathway (green overlay). Expression of this pathway can vary widely across genetically identical cells, as well as between different strain backgrounds. Here, turquoise (blue + green) and yellow (red + green) correspond to BC187 and YJM978 cells that have high GAL expression, whereas blue and red cells have low GAL expression. [Jue Wang, Renan Escalante-Chong]
Genome-Scale Metabolic Modeling
Jason Papin, Ph.D., an associate professor of biomedical engineering at the University of Virginia, is studying how to integrate high-throughput data into predictive, computational models for the interrogation of metabolic and regulatory networks. He is particularly interested in networks that relate to pathogens and human disease.
His lab recently compared the metabolic and virulence capabilities of two multidrug-resistant bacteria, Burkholderia cenocepacia and Burkholderia multivorans. Burkholderia are Gram-negative bacilli whose members run the gamut from potential bioterror agents to nonpathogenic species of interest in agricultural biotechnology.
“Multidrug-resistant pathogens are an enormous health concern as they can cause chronic infection in a variety of patients,” noted Dr. Papin, “and yet there are only limited sources for treatment. We studied two bacterial species that account for the majority of B. cepacia complex (BCC) infection in cystic fibrosis patients and other immunocompromised individuals.
“Although similar in their genetic compositions, they have important differences in pathogenesis. The predictive power of computational modeling and systems analysis provides a way to reconstruct the biochemical networks for hundreds to thousands of metabolites involved in virulence and other biological activities of pathogens such as these.”
Dr. Papin and colleagues utilized a variety of prediction/assessment tools to carry out the genome-wide reconstruction. These tools included flux balance analysis and the Model SEED, an online resource for the generation, optimization, curation, and analysis of genome-scale metabolic models.
“These models integrate annotated genome data and provide a framework for investigating the metabolic architecture of pathogens,” explained Dr. Papin. “The method accounts for all metabolic enzymes within a given cellular network, mapping the annotated genome to the proteome and associated chemical transformations in a mathematically driven comparative process.”
However, it’s not as simple as just pushing a button and then getting a cup of coffee. The analysis also requires human input. “I’d say 90% of the content of a network reconstruction requires 10% of the effort and time, but that last 10% of the content of the network reconstruction requires 90% of the overall effort and time,” advised Dr. Papin. “You can’t automate everything as it is still important for the scientist to carefully evaluate the currently available, and sometimes conflicting, data and literature.”
Overall, Dr. Papin’s team demonstrated by computational modeling that B. cenocepacia can produce a wider variety of virulence factors than B. multivorans and can utilize clinically relevant substrates more effectively while doing so.
“This supported the clinical observations that B. cenocepacia is more virulent,” remarked Dr. Papin. “Considering the large genomes and their ability for metabolic adaptation as a key factor in their pathogenesis, such reconstructions have wide-ranging implications for employing a systems biology approach for comparing the metabolic profiles of many other harmful pathogens as well as for developing new treatment strategies.”

The metabolic networks of multidrug-resistant Burkholderia pathogens are being characterized by University of Virginia scientists. Depicted here are the reactions common to both pathogens (gray) and the reactions unique to one pathogen or the other, namely B. cenocepacia (orange) or B. multivorans (blue). Models of the two reconciled metabolic networks enable prediction of the functional impacts of the differences in the enzymatic repertoire.
Stratifying Tumor Mutations
Cancers are complex (driven by a variety of genes), but also wildly heterogeneous, said Trey Ideker, Ph.D., a professor of genetics at the University of California, San Diego. “Gene combinations can vary greatly among patients,” he added. “Groups such as The Cancer Genome Atlas and the International Cancer Genome Consortium focus on systematically profiling tumors utilizing multiple levels of genome-scale data such as mRNA and microRNA expression profiles, DNA copy number and methylation signatures, and DNA sequences.”
Dr. Ideker and colleagues recently introduced a new way to analyze these complexities. With their approach, they hope to accomplish one of the fundamental goals of cancer informatics, the stratification of tumors into clinically and biologically relevant subtypes by comparing their molecular profiles.
“Although a promising new source of data to do this is the somatic mutation profile, which uses high-throughput sequencing of a patient’s tumor, this has proved challenging,” noted Dr. Ideker. “Fewer than 100 mutated bases can be found in most patient exomes and they are remarkably heterogeneous with often no more than a single mutation shared between clinically identical patients.”
The answer, he said, may lie in looking more broadly at combined data: “Cancer is not only a disease of single genes, but rather combinations of genes acting in molecular networks such as in cell proliferation and apoptosis. This suggests that cancers may share networks affected by these mutations.”
Although such cancer pathway maps are still being developed, researchers can access much relevant information from public databases of human protein-protein, functional, and pathway interactions. “By integrating these network databases with tumor molecular profiles, the molecular pathways of cancer are beginning to emerge,” Dr. Ideker indicated. “For example, we developed a network-based stratification method for integrating somatic tumor genomes with gene networks. This creates a stratification of various cancers into subtypes derived from clustering together patients who have mutations in similar network regions.”
In a proof-of-principal study, Dr. Ideker’s team used ovarian, lung, and uterine cancer cohorts from The Cancer Genome Atlas. The study identified network regions in each subtype that helped predict clinical outcomes such as patient survival time and development of drug resistance.
“We continue to improve upon the network-based stratification model,” Dr. Ideker asserted. “Although we demonstrated the utility of gene-gene interactions, other networks may be valuable to analyze such as those involved in signaling, metabolism, and transcription. We’ve only just begun to fully exploit the capabilities of using systems biology to stratify cancer subtypes.”
Mapping the Interactome
The genomic revolution is poised within our lifetime for an explosion that will provide a complete description of virtually all human genomic variations whether deleterious, advantageous, or neutral, suggested Marc Vidal, Ph.D., director of the Center for Cancer Systems Biology, Dana-Farber Cancer Institute. However, genomic sequencing alone cannot provide answers to fundamental questions relating to genotype-phenotype relationships. To “connect the dots,” proponents of the genomic revolution will have to decipher the complex cellular interactions of genes and their gene products. That is, they will have to explicate interactome networks.
Dr. Vidal and colleagues recently charted the proteomics of the human interactome network. Using a variety of systems biology tools, they generated a comprehensive binary protein-protein interaction map. They also plumbed the functional depths of thousands of proteins, as shown for candidates identified in genomic cancer screening studies. The investigators found specific interconnectivities between known and candidate cancer gene products.
“We created a new dataset experimentally of 14,000 interactions and compared it to existing literature datasets totaling 11,000 interactions,” said Dr. Vidal. “The main message is that at this point we have mapped more interactions than the whole community combined, and our dataset is much less systematically biased.”
As systems biology continues to make strides in technology, modeling, and deciphering complex biological interactions, immediate benefits include identifying disease-related mechanisms and biomarkers, and revealing novel drug targets.

