
December 1, 2009 (Vol. 29, No. 21)
Gene Cloning and Protein Expression in Insect Cells Is Faciliatated by New Methodologies
Since the pioneering interferon expression studies conducted in the early 1980s, baculovirus-based expression in insect cells has become one of the most widely used systems for production of recombinant proteins. Several unique features account for the popularity of this approach.
- It is a eukaryotic expression system, therefore it uses many of the protein modification, processing, and transport pathways present in higher eukaryotic cells.
- High expression levels (up to 1 g of protein product/L of culture) can be obtained, without many of the hassles encountered when using mammalian cell cultures.
- The viral genome can accommodate relatively large fragments of foreign DNA.
- The virus can be propagated to high titers without the need of a helper DNA.
- Availability of suspension cell lines (many of them adapted to grow in serum-free conditions) makes the system amenable to scale-up.
- As part of the baculoviridae family, the Autographa californica multiple nuclear polyhedrosis virus (AcMNPV) is safe to use as it is noninfectious to vertebrates.
- A wide variety of transfer vectors have been developed that made the recombinant virus isolation a simple process.
A variety of approaches to transfer genes from standard vectors into the viral genome have been developed. Efforts to ease the requirement for a plaque assay resulted in the design of a bacterial transposition method marketed under the name of Bac-to-Bac®, from Invitrogen, part of Life Technologies. Concurrently, an in vivo recombination method between a transfer vector and a restricted viral DNA was developed.
The approach, which restores the function of an essential gene, was further improved to include a blue vs. white plaque visualization method, and it is currently commercialized under the name of Bac-N-Blue™.
More recently an in vitro recombination system has been developed. In this system (commercially available under the trademark of BaculoDirect™) the foreign gene is transferred from an entry clone to the viral DNA via Gateway LR recombination. The thymidine kinase gene present in the Gateway® cassette acts as a strong counter selectable marker when cells are grown in the presence of ganciclovir.
In this article, we present three new pFastBac™/TOPO® vectors, compatible with the Bac-to-Bac system, that allow the expression of poly-histidine protein fusions either cell-associated or secreted into the medium. We also generated a Gateway-compatible BaculoDirect genome that enables the expression of intracellular N-terminal GST protein fusions.
Polyhistidine Fusion Proteins
The topoisomerase-adapted vectors pFastBac/NT-TOPO and pFastBac/CT-TOPO (Figure 1A) allow the production of N-terminal and C-terminal intracellular polyhistidine fusion proteins. Blunt-ended PCR products can be cloned with an efficiency >95%. To test the expression performance of the vectors, several human genes were cloned and eventually expressed in SF21 cells. All the proteins tested could be readily detected as sharp bands with the expected molecular weight in Western blots. It is worth noting that higher yields were consistently observed in those samples derived from pFastBac/NT-TOPO.
A subset of the samples was subjected to TEV protease cleavage. Even when lysates, rather than purified proteins, were employed, protease digestion resulted in virtual completion in only 30 minutes of incubation (Figure 1B).
To further validate the performance of these new type of vectors, a notoriously difficult-to-express protein, the serotonin GPCR 5HT1a was cloned into pFastBac/CT-TOPO and the resulting viral particles were used to infect High Five cells. The 5HT1a protein localized primarily to the surface of the cells, and the protein was properly folded as suggested by the corresponding ligand binding assay (Figure 1C).
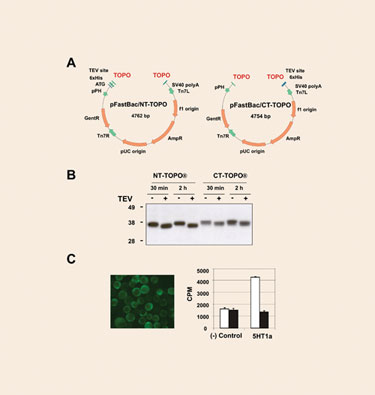
Figure 1. New pFastBac/TOPO vectors for intracellular expression
Extracellular Secretion
In order to provide a suitable expression platform for those proteins that require post-translational modifications such as glycosylation or disulfide bond formation, we generated pFastBac/HBM-CT-TOPO (Figure 2A)—a vector that facilitates the translocation of the cloned gene products into the endoplasmic reticulum. The vector harbors the Honey Bee Melittin (HBM) secretion signal coding sequence, which has been placed next to the topoisomerase binding site, upstream of the gene of interest (Figure 2A).
To validate the effect of the HBM signal sequence, we first generated recombinant expression baculovirus particles by cloning the emerald green fluorescent protein (EmGFP) gene into pFastBac/HBM-CT-TOPO (pFastBac/CT-TOPO and pFastBac/ NT-TOPO were used as controls), and used the generated particles to infect SF21 insect cells.
EmGFP with a HBM secretion signal sequence had a distinguished localization pattern compared to N- or C-terminal his-tagged EmGFP which was evenly distributed throughout the cytoplasm and nucleus in infected cells. The HBM-EmGFP fusion protein localized primarily in the periphery area of the cells and had a granular appearance, suggesting that the protein is in the process of secretion (Figure 2B).
To confirm this hypothesis, cellular and extracellular fractions were analyzed by Western blots. Although EmGFP from pFastBac/HBM-CT-TOPO and pFastBac/ CT-TOPO expressed at comparable levels (Figure 2C, lanes 3 and 4), only EmGFP expressed from pFastBac/HBM-CT-TOPO was found on the medium (Figure 2C, lanes 7 and 8), confirming that the HBM secretion signal sequence is able to promote secretion EmGFP. On a side note, the EmGFP levels produced by pFastBac/NT-TOPO were so high that leakage into the extracellular medium was unavoidable (Figure 2C, lanes 1 and 6).
To assess the relative secretion efficiency of proteins fused to the HBM signal sequence, we replaced the original signal sequence of two ordinarily secreted proteins by the HBM sequence, and we examined the presence of these protein products in the extracellular medium. We used human coagulation factor IX (F9) and erythropoietin (EPO) as models. Their corresponding genes were cloned into pFastBac/CT-TOPO (including their natural signal sequence coding region) and into pFastBac/HBM-CT-TOPO (excluding their signal sequence coding region).
Four different proteins were, therefore, expressed: wild type F9, wild type EPO, and their corresponding mature peptides fused to the HBM signal sequence. Virus particles were obtained and used to infect SF21 and High Five cells. Cell culture media were collected 24h, 48h, and 72h post-infection, and the secreted proteins were analyzed by Western blot.
Proteins were secreted to comparable levels regardless of what signal sequence the protein was fused to (Figure 2D). The relative electrophoretic mobility of the different proteins suggests that the signal sequence was correctly cleaved (Figure 2D). The amount of EPO and F9 in the culture media increased up to 8 and 2 mg/L respectively after three days post-infection. The results confirm that the HBM signal sequence can function as efficiently as the wild type signal sequences of EPO and F9 to promote the secretion of proteins validating the yeast-derived HBM signal peptide as an efficient protein secretion vehicle in insect cells.
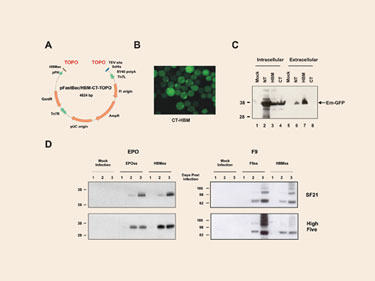
Figure 2. Performance of pFastBac/HBM-CT-TOPO
Expression of GST Fusion Proteins
We generated a Gateway destination vector based on the AcMNPV genome, which allows the production of N-terminal GST-tagged protein fusions (Figure 3A). The Gateway cassette harbors the counterselectable thymidine kinase (tk) and ß-galactosidase (lacZ) genes. An aliquot of a Gateway LR reaction is directly used to transfect insect cells, which are grown under gancyclovir selection. Recombinants must exhibit a white appearance when cells are (optionally) plated on X-gal-containing solid medium, confirming the loss of the lacZ gene.
To test the performance of the vector, the EmGFP coding sequence was transferred from a Gateway entry clone to the baculoviral genome, which was then transfected into SF21 cells. The corresponding P2 viral stock was used to infect SF21 cells and to assess GFP expression (Figure 3B). Expression was confirmed by Western blot (Figure 3C).
The new vectors presented in this study enable flexible and rapid gene cloning and protein expression in insect cells. The vectors allow the production of proteins tagged with poly-histidine motif (either at the N- or C-terminal regions) and with GST (at the N-terminal end), facilitating their purification. The presence of a TEV protease recognition site permits the generation of nearly native proteins leaving as few as two or six additional amino acids (pFastBac/NT-TOPO and pFastBac/CT-TOPO respectively).
Completely native proteins (nontagged) can be also obtained by using the pFastBac/CT-TOPO vector and including a stop codon at the end of the gene sequence. The vectors are adapted to the TOPO or Gateway technologies allowing fast, efficient, and restriction enzyme independent cloning.
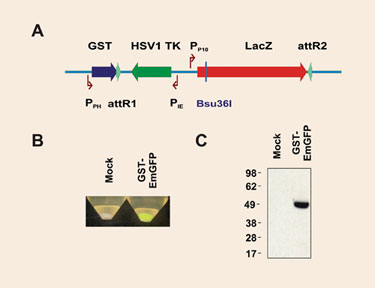
Figure 3. Performance of the BaculoDirect GST vector
Jian-Ping Yang, Ph.D. ([email protected]), Federico Katzen, Ph.D., and Sanjay Vasu, Ph.D., are senior scientists, Lansha Peng is a senior associate scientist, and Wieslaw Kudlicki, Ph.D., is a research fellow in R&D at Life Technologies. Web: www.lifetechnologies.com.

