Progress in all of the areas of biologics production has always been incremental and not marked by sweeping breakthroughs. The fine points of the latest hard-won advances in bioprocessing technology and operations were presented and debated at IBC’s “Early to Late Stage Bioprocessing Conference,” which took place in Boston.
Biologic drugs are a special category with regard to their patentability and resistance to patent infringement, according to Joan Shankle, managing director of Aurum Group. They are often complex mixtures, there are limits to the ability to characterize and identify their structures, the active components of the molecule may not be fully identified, and they are difficult to manufacture consistently.
For these reasons regulatory guidelines need to address product comparability rather than identity, as is the case with small molecule drugs. The latter are covered by the Hatch-Waxman Act, since small molecules can be chemically synthesized and identical versions produced.
“Historically, there was a fork in the regulatory road that occurred around 1993 between biologics and well-characterized biotechnology products. At this time the latter moved from CBER (Center for Biologics Evaluation and Research) to CDER (Center for Drug Evaluation and Research), and in 1996 the comparability guidance was issued,” Shankle continued.
“The basis for the differentiation was the well-characterized designation resulting from improved manufacturing (more consistent and better control) and also improved analytical techniques that facilitated a detailed description of protein structure, composition and function,” she added.
According to Shankle, the 1996 FDA Comparability Guidance ruling addressed the issue, taking into account improvements in technology for the characterization of macromolecules. This allowed more flexibility in manufacturing facilities and processes. “The most important assessment factor for FDA is whether manufacturing changes translate into significant differences in safety or efficacy,” she said.
Comparability testing demands that manufacturers should carefully assess modifications and evaluate the resulting molecule for similarity to the preexisting product through a side-by-side analysis. Shankle advised that manufacturers should take advantage of the benefits and flexibility provided by current comparability regulations, investing in the analytical tools necessary to identify critical attributes of the molecule early in the process, thus allowing the monitoring of product quality through all phases of development.
Shankle’s comments on biosimilar products call to mind ongoing discussions and proposed legislation regarding biogenerics. As the technology for characterization has developed, and as the patents for a number of biologic drugs are running out, the question has become more pressing. The EU has moved ahead of the U.S. by establishing a new system for evaluation of biological medicinal products that assumes that bioequivalence is not sufficient for approval but rather additional studies will be required.
In the EU legislation, substantial nonclinical and clinical data are now mandatory. Marketing authorization applications are submitted to The European Medicines Agency and reviewed by the Committee for Medicinal Products for Human Use. Canada has also fallen into line with similar legislation.
Currently in the U.S. Congress, the Waxman-Clinton-Schumer bill is under consideration, and while stalled for some time, it is expected to move ahead as the new administration puts forth a comprehensive healthcare overhaul. BIO has voiced concern over the legislation, which will, no doubt, be a contentious issue in the upcoming congressional session. Nonetheless, considering the staggering cost of biopharmaceuticals, there is a pressing need to develop a coherent regulatory process to allow biogenerics to obtain approval in the U.S.
For many companies, bioprocessing of a product may involve outsourcing one or more steps. Paul Mehelic, Ph.D., principal scientist and group leader at Pfizer, discussed solutions to challenges that arise in the course of an antibody-production project. These include failure of the scale-up phase to meet expectations, disagreements between client and contract manufacturer organization (CMO), changes in personnel midway through the project, and platform changes or modifications during the campaign.
Adventures in Outsourcing
In the program that Dr. Mehelic described, the pilot batch of a monoclonal antibody produced by the contract manufacturing organization fell to a low purity level, including a high amount of aggregates, excessive host cell protein, and various other unacceptable impurities. In addition, there was a crash of the cell population at 340 hours, despite adherence to the prescribed feeding schedule. In gel separations, atypical bands and other singularities were observed.
At this point, Dr. Mehelic’s team was faced with the decision to either proceed with evaluating the pilot batch or delay the program until a new initial lot had been prepared. Proceeding with the original pilot batch could save time, but could result in failure if it did not meet purity and stability criteria. Because of the substantial risk, the decision was made to delay GMP production for six months until the problem involved in the purification process could be resolved.
With the recommendations of the Mehelic team, the CMO made a number of changes in the protocol, resulting in significant process optimization. These included earlier harvest time, changes in protein A chromatography conditions, modifications in diafiltration, and alterations in CEX chromatography. With these changes in place, the project proceeded with production of the GMP batch, which is now in clinical trials.
“In the final analysis we let the data drive the discussions and the final decision making process,” Dr. Mehelic concluded.
Clone Selection
“Look closely at what you do early on in process development, because you’ll have to live with those choices at the late stage,” say W. Blaine Stine, Ph.D., associate research investigator at Abbott Laboratories.
Frequently, teams working at the bench seek to optimize activity and minimize toxicity, while being oblivious to physicochemical parameters such as protein solubility and stability. So, Dr. Stine laid out a case study detailing how to manage these features at an early stage during the process of clonal selection after the introduction of back mutations, so as to obtain the overall optimal outcome. This investigation comprised nine IgG antibody-producing clones that had been modified by the introduction of select back mutations into the framework regions of both the light and heavy chains.
Dr. Stine’s tools for characterization include capillary isoelectric focusing, capillary zonal electrophoresis, size exclusion chromatography and differential scanning calorimetry.
The group determined that all nine clones have similar charge isoform distribution, and like-for-like amino acid substitutions did not affect the isoform distributions, while neutral for-charged substitutions resulted in predictable shifts in the entire isoform population. This was apparent for substitutions as small as a single charged amino acid in a protein comprised of over 1,400 residues.
Dr. Stine’s group performed size exclusion chromatography studies to measure the propensity of the molecules to aggregate and fragment and differential scanning calorimetry studies to measure the thermodynamic stability on the nine target clones. The rank order stability determined using the two approaches was shown to be in agreement. Correlating the stability data from the nine clones with the corresponding changes to their amino acid sequence revealed that substitutions at three specific positions appear to affect thermodynamic and accelerated stability.
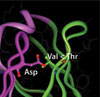
To better understand this relationship at a mechanistic level, they turned their attention to molecular modeling for the theoretical vindication of their experimental results. The computer modeling results identified three locations where substitutions affected stability. Two of these locations involve a pair of amino acids with closely interacting side chains. The methionine-valine pair was more stable than the isoleucine-alanine pair, which is consistent with the entropic gain from the increase in the number of rotational bonds. The third location was shown to closely interact with an aspartic acid residue.
Experimental data demonstrated that threonine was more stable than valine at that location. This difference is consistent with a molecular model suggesting an unfavorable polar/non-polar interaction with valine present, whereas with threonine there is the potential for the formation of a stabilizing hydrogen bond with the aspartic acid side chain.
These examples support Dr. Stine’s contention that the modeling allows one to develop a mechanistic understanding of how energetically favored substitutions relate with observed changes in physicochemical stability. Moreover, they have the very practical consequence of allowing the investigator to predict the changes that could stabilize a clone which may have favorable binding properties, but would otherwise be excluded because of its molecular fragility.
Improved Protein Expression
“Our Paveway technology optimizes three elements: vectors, host strains, and fermentation protocols,” stated Andy Topping, director of early-phase development at Avecia Biologics. Topping profiled the company’s strategies for maximizing titers of recombinant proteins and reducing development time and costs with the aim of building more effective manufacturing processes.
In order to build more efficient vectors, the company has examined the control elements in order to improve their performance. DNA looping is an important key to improved production because of its ability to foster cooperativity between binding sites on the DNA. It is generated by proteins or complexes of proteins binding to two different regions. When the appropriate operator genes are placed next to the target structural genes, a hair-pin effect is produced in which the repressor protein pulls together the two operators, and a loop of thousands of base pairs of DNA bulges out.
So the simultaneous binding of the two sites drives protein production down to zero. Addition of the appropriate inducer releases the repressor and allows a burst of protein synthesis. Using this approach, protein production as high as 25% of total cell protein was observed and moreover could be tightly controlled with slower expression, better folding, and slow accumulation for many hours with continued cell growth.
With respect to host strain selection, from a panel of E. coli candidate strains, those that displayed the best expression of recombinant proteins and the most robust fermentation performance at high cell densities were chosen. This left the fermentation platform as the third target for optimization. Avecia has made numerous changes including the use of preestablished cell bank protocols in which the strain history and other inputs are well known, and a scalable feed strategy predeveloped for high protein productive biomass levels.
With all these improvements in place, the company was able to achieve high titers of protein production, in some cases higher than 14 grams per liter. However, such high levels of productivity require gargantuan quantities of resin, as much as 400 L for protein purification. So clearly the benefits of increased productivity cannot be realized unless the downstream bottleneck can be removed.
To come to grips with this impasse, the company has developed a high-yield/high-capacity generic purification approach for antibody purification that consists of clarification of the culture supernatant, protein A affinity columns, and final polishing steps with AIEX resin (anion exchange resin) combined with CHT (ceramic hydroxyapatite).
“We then asked, is there a comparable generic scheme for antibody fragment purification?” Topping explained.

Since antibody fragments lack an Fc region (the binding portion to protein A), it was necessary to search alternative synthetic affinity resins. The Avecia team carried out a rapid screening program of many different resins and identified mixed mode cation exchange resins as being particularly promising. This proved to be quite satisfactory, although cleaning, resin capacity, and possible ligand leakage are all concerns.
Topping argues that the Paveway technology achieves tight control with effective modulation of expression for a range of recombinant proteins. “We are able to progress rapidly from gene to high titer fermenters,” he stated, “and we now have a proven track record with many antibody fragments.”
Developmental Black Holes?
“Frequently, investigative drug research projects become mired down when their management is not properly integrated,” stated Mark Cunningham, Ph.D., drug discovery researcher at Centocor. The company’s decision to form cross-functioning teams, referred to as pharmaceutical development advisory committees (PDAC), was based on a monoclonal antibody development project that became bogged down when standard optimization protocols failed to produced the desired results.
Introduction of mutations into the CDR region of the antibody, while they improved performance, resulted in low expression levels and precipitate formation, and after a year the project was abandoned. So the formation of a multidisciplinary team was conceived of as a means of overcoming roadblocks early on, and moving to a successful conclusion.
“The primary aim of the PDAC is to act as advisory committee, guiding the research forward,” Dr. Cunningham explained. “This means that the discovery teams present their findings on the biophysical properties of candidate molecules, and the interaction provides guidance to move the work forward through any issues posed by the investigation.”
As the process has been in effect since 2005, it has accumulated a considerable track record, and at this time four monoclonal antibodies and two other biologics are working their way through to the goal at which an optimized lead compound is identified.
In one case, the existence of labile methionine residues in the CDR was identified as a source of chemical instability. However, since replacing all the methionines resulted in a loss of affinity, the team recommended a stress analysis of the molecule to determine which replacements would be the most satisfactory.
In another case study, a biologic displayed excessive cleavage and aggregation. The recommendation was to redesign the molecule with a glycosylation site and an engineered peptide sequence, thereby overcoming these issues.
Dr. Cunningham emphasized the ongoing nature of the pharmaceutical design advisory committees. “This process incorporates quality by design attributes, and is constantly evolving,” he emphasized.
The vast complexity of developmental pathways in biologic product development calls for the management skills of Solomon, but this in turn requires suitable technology, otherwise the baby may be cut in half without benefit to the advancement of the project.
One theme that has been prominent in this and other bioprocessing conferences is the need to make decisions early on that endure through upscaling and clinical trials. Yet this admonition conflicts with the need for quick and dirty in the early stages. Even with the wisdom of Solomon, experience, acute judgment, and luck are critical factors.
