
February 1, 2013 (Vol. 33, No. 3)
Maintaining Disulfide Bridges Leads to Improved Homogeneity and Stability for ADCs
A new wave of antibody-targeted anticancer therapies is showing great clinical promise, with the potential to transform cancer treatment. These antibody-drug conjugates (ADCs) are multicomponent systems that combine recombinant and chemical technologies to deliver a highly potent cytotoxic drug to a tumor thereby minimizing off-target toxicities and increasing the therapeutic window of the drug.
While there are multiple chemical strategies to conjugate a payload to an antibody, one of the more established methods utilizes the cysteine thiols derived from a reduced interchain disulfide bond. The linkers that are currently used conjugate only to a single cysteine thiol, which results in the loss of covalency between the cysteines that composed the original disulfide.
Reduction of the four interchain disulfides of a standard IgG1 antibody gives rise to up to eight thiols, so when a drug is conjugated a range of positional isomers with drug to antibody ratios (DAR) ranging from 0 to 8 are produced. Each drug positional isomer may have its own distinct biological profile, and it is difficult to control the conjugation reaction to achieve the required DAR.
If the disulfide can be re-bridged following conjugation, there will be fewer possible drug positional isomers than with the single thiol approach, with the DAR distribution limited to between 0 and 4 and a DAR between 2 and 4 achievable at greater than 94%.
In this article, a conjugation technology called ThioBridge™ from PolyTherics is described that allows site-specific targeting of disulfide bonds in a way that retains the covalent integrity of a disulfide bridge and does not require re-engineering of the antibody to obtain a less heterogeneous and stable ADC.
While great progress has been made in the selection and optimization of the antibody and cytotoxic payload, many of the ADCs in development are typically heterogeneous mixtures possessing populations with varying DARs. Producing a more homogeneous ADC with the optimal DAR distribution for safety and efficacy is challenging with current conjugation methodologies, and it is widely recognized that further improvements are needed if next-generation ADCs are to really fulfill their potential.
The development of a successful ADC is predicated on appropriate target selection, as well as optimization of the antibody, the linker, and the cytotoxic agent. While much attention is given to the linker in terms of its ability to release the payload after it reaches the target cell, less attention is given to the fact that the linker also has to serve to conjugate the payload as efficiently and as selectively as possible to the antibody.
Although high DARs, i.e., above 4, are observed to be more potent in vitro, higher loaded species are more hydrophobic due to the nature of the drug payload, with increased clearance rates and in vivo toxicity reported for these species. There is also increasing concern about the in vivo instability of ADCs due to the deconjugation of maleimide-based linkers from the antibody while in circulation. Such instability can cause the cytotoxic drug to be released before it reaches the tumor, potentially leading to off-target toxicity and reduced efficacy.
Once released, the cytotoxic payload can potentially conjugate to serum proteins and thus remain in the circulation rather than reaching the tumor.
Utilizing the latent reactivity of a reduced disulfide for conjugation avoids the need to re-engineer the antibody specifically for site-specific conjugation and is therefore more generally applicable to a wider range of antibodies. While antibodies possess many disulfide bonds, only the heavy to heavy and heavy to light interchain disulfides are both reducible and accessible for conjugation under mild conditions. These interchain disulfides are easily reduced either fully or partially using standard reductants.
Reagent Usage
The reduction of an accessible disulfide bond leads to the generation of two free cysteine thiols, and these can be site-specifically conjugated with a bis-thiol alkylating reagent (ThioBridge) to which the payload is already conjugated via a releasable/nonreleasable linker.
The ThioBridge disulfide-bridging reagent undergoes bis-alkylation to link to both cysteine thiols derived from the reduced disulfide (Figure 1). The reagent is capable of undergoing interactive Michael and retro-Michael reactions to allow the thermodynamic product to be formed. This enables the two free thiols to re-anneal across a 3-carbon bridge, which is small enough and flexible enough to not cause disruption of the protein’s tertiary structure, which is otherwise primarily stabilized by noncovalent interactions.
Mechanistically, an alpha-beta unsaturated double bond in the ThioBridge reagent is required to initiate a sequence of addition-elimination reactions. Once thiolate addition occurs, elimination of the remaining sulfone moiety becomes possible as a sulfinic acid moiety. This generates another conjugated double bond for the addition of a second thiolate anion and the formation of a 3-carbon bridge between the two sulfur atoms. The end result is two very stable thiol-ether bonds in place of the original sulfur-to-sulfur disulfide bond.
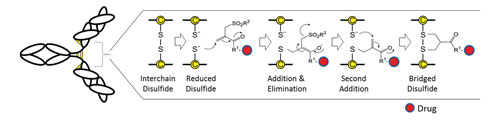
Figure 1. Drugs can be conjugated via the interchain disulfides of an antibody using ThioBridge, which maintains the covalency of the original disulfide by re-bridging the two sulfur atoms via a sequence of addition and elimination reactions.
ThioBridge can be used with a wide range of payloads and cleavable linkers. ThioBridge reagents and the processes for conjugation are straightforward and tolerant of a wide range of reaction conditions, such as pH and antibody concentration. The antibody is first incubated with a disulfide reducing agent such as dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP) for either partial or full reduction of the interchain disulfides. Typical conditions are 2 hours at 37°C with 1–2 equivalents of TCEP per disulfide. If the reductant is used in excess then a buffer exchange step may be required to remove the residual reductant.
The process (Figure 2) is similar at this point to a maleimide-based approach. The ThioBridge reagent is then added to the reduced antibody, and the mixture is incubated for 2 to 5 hours at ambient temperature. SDS-PAGE can be used to analyze the reaction mixture since re-bridged disulfides will not lead to interchain dissociation due to the SDS (e.g., Figure 2).
A diafiltration step can be used to remove excess reagent and buffer exchange to a final storage buffer. The resulting ADC can then be analyzed by chromatographic methods, such as by hydrophobic interaction chromatography, to determine DAR.
This disulfide-bridging conjugation has been used successfully to conjugate biocompatible polymers for serum half-life extension for many different types of proteins including interferons, blood factors, enzymes, and antibody fragments.
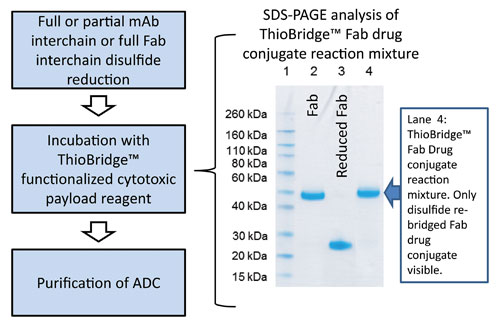
Figure 2. Process flow for creating an ADC based on a ThioBridge antibody conjugation linker: The key step is the reduction (full or partial) of the antibody interchain disulfides prior to adding the ThioBridge cytotoxic payload reagent. Also shown is SDS-PAGE analysis of a ThioBridge Fab drug conjugate reaction mixture. Since unbridged Fab runs as separate heavy and light chains, the single higher MW product band demonstrates the re-bridging of the interchain disulfide with ThioBridge.
The ThioBridge process is particularly suited to creating drug conjugates based on Fabs, which have a single accessible disulfide distal to the Fab’s binding region. A homogeneous Fab drug conjugate with a DAR of 1 is obtained (Figures 2 and 3). There is some evidence that Fab drug conjugates are better able to penetrate the tumor mass than whole antibodies due to their smaller size. However, since Fabs do not contain an Fc region to extend serum half-life by FcRn-mediated recycling, some pharmacokinetic optimization may be required. Since ThioBridge incorporates a hydrophilic polyethylene glycol (PEG) spacer, half-life can be readily optimized by controlling the size of the PEG spacer.
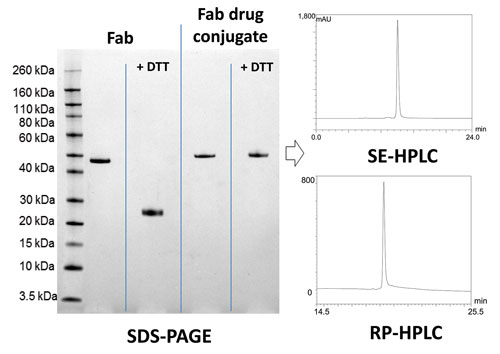
Figure 3. Analysis of a ThioBridge Fab drug conjugate by SDS-PAGE, SE-HPLC, and RP-HPLC: Since Fabs have a single interchain disulfide, a single DAR species (i.e., 1) is produced. Incubation of the Fab drug conjugate with DTT does not lead to competitive deconjugation of the payload using ThioBridge, demonstrating the excellent stability of the approach.
Conclusion
The ThioBridge conjugation process is stoichiometrically efficient, typically requiring no more than 1.5 molar equivalents of ThioBridge reagent to be used per disulfide. The bridging approach also at least halves the requirement for cytotoxic reagent compared with single thiol reactive reagents, since the reactive sites are halved. This advantage helps to reduce the cost of the ADC production and allows for a less complex purification process.
Antony Godwin, Ph.D. ([email protected]), is director of science & technology at PolyTherics. The author is grateful to the ThioBridge project team at PolyTherics and to George Badescu, Ph.D., head of bioconjugation & bioanalysis, for his assistance in preparing the tutorial.

