
December 1, 2011 (Vol. 31, No. 21)
Investigations Are Beginning to Elicit More Selective and Specific Druggable Targets
How is the current model of drug discovery faring? “It’s not working,” stated Chas Bountra, Ph.D., chief scientist at the Structural Genomics Consortium (SGC) and professor of translational medicine, University of Oxford. “There are too many targets to choose from, and many of the targets being worked on today will not deliver therapeutically because they are at too late a stage in the disease cascade.”
Dr. Bountra was speaking at the recent “Epigenetics for Drug Discovery” conference, which was organized by Cellzome, CellCentric, and BioFocus, all companies with interests in the field of epigenetics.
Tony Kouzarides, Ph.D., deputy director of the Gurdon Institute, University of Cambridge, agreed. “Much drug discovery has hinged on targeting kinases found in the cytoplasm. Perhaps by getting a crucial hit on a molecule in the nucleus, we might find better druggable targets.”
Both speakers were suggesting that by studying the epigenetic information superimposed on the genome, such as modifications to the histone proteins that package DNA in chromatin, it may be possible to identify and more effectively block some of the cascades that cause disease states to occur.
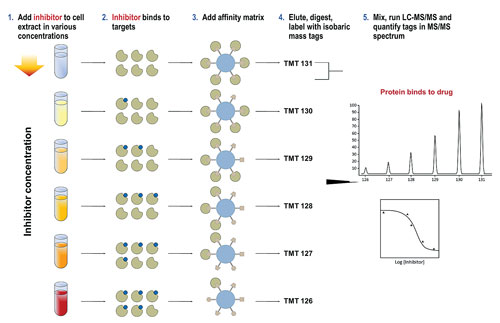
Cellzome has developed a different kind of cell-based assay using native proteins.
A Safe BET?
One family of epigenetic-derived drug targets that dominated discussion at the conference were the BET (bromodomain and extra-terminal) proteins, a family of four proteins that selectively recognize and bind to acetylated lysine residues in histone. BET proteins stimulate transcription by recruiting specific types of proteins, for example the super elongation complex (SEC), to chromatin, leading to stimulation of transcriptional elongation of certain target genes including oncogenes and pro-inflammatory cytokines.
As a consequence, targeting BET proteins and displacing them could provide a method of inducing cell cycle arrest and even cell death of defectively programmed cells. This is why inhibiting the action of BET proteins is being developed as an interesting strategy for treatment of cancer and inflammation.
The BET proteins BRD4 and BRD3 have been implicated as part of the disease cascade for mixed lineage leukemia (MLL), an aggressive form of cancer that is hard to treat and mainly affects infants.
“Proteomics evidence shows the SEC, and by inference MLL-fusions, are associated with BET proteins,” said Dr. Kouzarides. “Working with Cellzome and GlaxoSmithKline (GSK), we have characterized a small molecule we call I-BET 151 that interacts with BRD4 and BRD3 BET proteins and displaces them from genes regulated by MLL-fusions.”
Dr. Kouzarides presented data to show that following treatment with I-BET 151 on two different human MLL-fusion cell lines (MV4;11 and MLL-AF9), there was a clear apoptotic effect seen, and transcription of key oncogenes BCL2, C-MYC, and CDK6 was repressed.
Additionally, in animal models, mice transplanted with MV4;11 cells and treated with I-BET 151 survived without disease, and those transplanted with MV4;11 cells and not treated with I-BET 151 all died within 30–40 days. In a second animal model, mice transplanted with MLL-AF9 cells and treated with I-BET 151 all survived free of disease, and again the untreated mice died within 12–13 days.
“We hope to progress I-BET 151 into clinical trials soon because we have shown that it kills leukemic cells in mice by displacing BET proteins from chromatin,” Dr. Kouzarides concluded. “Therefore, this has the potential to become the first potent and selective epigenetic drug.”
NUT midline carcinoma is another form of cancer in which BET proteins have been implicated. Stefan Knapp, Ph.D., from the University of Oxford explained, “NUT midline carcinoma is a rare, aggressive human cancer in which most of the coding sequence of NUT is fused with the BET protein, BRD4 or BRD3.
“In collaboration with the Dana Farber Cancer Institute, we have synthesized a small molecule based on available patent literature from Mitsubishi and GSK that we call JQ1, that selectively inhibits the BET protein BRD4.”
Dr. Knapp presented data to show that following JQ1 treatment of mice with xenografts of cells from a patient’s NUT midline carcinoma chest tumor, apoptosis occurred and the tumor was reduced in weight and volume.
“BET proteins are now becoming high profile as druggable targets,” Dr. Knapp summarized. “Around 20 percent of tumor cell lines respond well to BET inhibition, and there are many chemotypes we have identified that bind to BRD4, so there is plenty of opportunity to develop small molecule inhibitors.
“Based on recent results on BET inhibition in cancer, a spin-off company from the Dana-Farber Institute, Tensha Therapeutics has already been set up to take JQ1 analogues into the clinic and has recently received initial funding of $15 million,” he added.
Soren Beinke, Ph.D., investigator in the Epinova DPU at GSK, also presented on BET inhibition, showing that the I-BET small molecule could be used as an anti-inflammatory agent.
Dr. Beinke’s group, along with a team at the Rockefeller Institute, demonstrated that I-BET suppressed the expression of key inflammatory mediators in macrophages. I-BET also prevented endotoxic shock in mice dosed with bacterial lipopolysaccharide, and attenuated bacterial induced sepsis.
Most promisingly for therapeutic applications, a single dose of I-BET applied after lipopolysaccharide injection, at the time when mice started to develop symptoms of inflammatory disease, cured the mice. “I-BET could be a good candidate for the effective suppression of acute inflammatory conditions,” Dr. Beinke suggested.
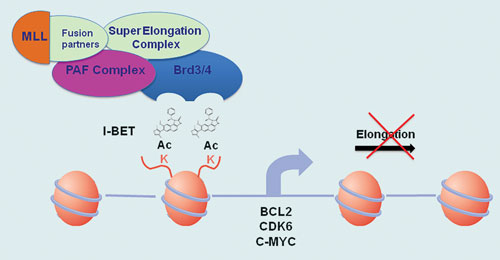
Epigenetic action of I-BET inside the nucleus of MLL cells [University of Cambridge]
Another Promising Target?
Enzymes such as histone deacetylases (HDACs) or histone methyltransferases (HMTs) have for some time represented potentially druggable epigenetic targets. BioFocus discussed its work on HDAC inhibitors for treating Huntington disease.
Roland Bürli, Ph.D., senior director scientific alliances at Biofocus, believes that a poly Q extension at the N-terminus of the huntingtin protein is thought to play a central role because the length of the poly Q repeat inversely correlates with Huntington disease onset.
“Working with CHDI, a not-for-profit organization dedicated to finding therapies to treat Huntington disease, we have developed HDAC4 inhibitors and tested them in multiple biochemical and cell-based assays,” Dr. Bürli commented. “The most promising lead compounds are currently being scaled up for further studies in rodents.”
Current and Future Challenges
As well as proving efficacy of epigenetic-derived small molecules, there are other technical and commercial barriers to successfully identifying and making progress with these types of drugs.
One issue of working with epigenetic targets was identified by Gerard Drewes, Ph.D., vp of discovery research at Cellzome. According to Dr. Drewes, epigenetic processes are controlled by multiprotein complexes such as HDACs, and these complexes are difficult to recreate when using recombinant proteins in binding studies.
“HDAC cell-based and biochemical assay data is not always consistent, as it can be difficult to assess the activity of a compound when using it on an isolated recombinant protein. Typically, recombinant proteins or enzymes don’t have the same conformation and activity as a protein in a protein complex. To overcome this problem, we developed a different technique using native proteins for the assay, which we call chemoproteomics.”
The initial step in Cellzome’s approach is similar to affinity chromatography, in that it uses a bead matrix to capture different proteins, including many epigenetic enzymes, in their native form. Compounds are added to cells or tissue (either in vitro or in vivo), which then compete with the ligands of the bead matrix for binding of enzyme targets and their complexes. Subsequently, this competition is quantified using mass spectrometry, and a profile of target proteins is determined.
Dr. Drewes’ data showed that when his group profiled a panel of 20 HDAC small molecule inhibitors including known HDAC inhibitor drugs such as SAHA (Zolinza/Merck & Co.) and romidepsin (Istodax/Celgene), IC50 curves for protein subunits could be measured and both known and novel protein targets identified.
“We have established the utility of the chemoproteomics technology for HDAC inhibitors,” Dr. Drewes said. “The advantages of using this method is that it is quicker than the standard recombinant workflow and represents a highly multiplexed assay where the IC50 of a compound for 100 or more proteins can be measured in one run.
“This is essentially an unbiased technology that allows the study of the ‘chemical space’ in which HDAC inhibitors operate, and it is this that will help us discover more novel protein targets.” Dr. Drewes also noted that the same technology is already being applied to other classes of epigenetic targets.
Dr. Bountra highlighted a more far-reaching challenge, which has dogged the pharma industry and could hinder progress in epigenetic drug discovery. “All pharmas and biotechs are working on the same targets in parallel, in secret, so there is a massive duplication of effort.”
“The tragedy of drug discovery attrition figures is that they are also duplicated and the outcomes are not shared. This means thousands of patients are exposed to molecules that many other groups definitely know are destined to fail in trials. That cannot be ethically or morally right. If we’re to make significant rapid progress with epigenetic drug discovery, this situation has to change.”
According to Dr. Bountra, one problem is that early IP on molecules means information is not shared, which makes drug discovery slower, more difficult, and more expensive. The solution, he believes, is to establish public private partnerships such as the SGC, which includes academic collaborators in the U.K., U.S., Canada, and Sweden, alongside pharma companies such as GSK, Novartis, and Pfizer.
These partnerships would take industry standard molecules for novel (“pioneer”) targets through to Phase II proof of clinical mechanism, without having any IP on the molecule. This would enable rapid publication of all data including negative clinical results, and would also de-risk the process, as novel targets would be clinically validated before large, parallel investments in multiple organizations.
Dr. Bountra cited the epigenetic-derived molecule JQ1 as an example of how this could work. “JQ1 has been given freely to more than 100 labs worldwide, and is now being profiled in several therapy areas including inflammation, virology, and some forms of cancer.
“The SGC has demonstrated that having the reagents freely available accelerates collaboration, and therefore science, and hence drug discovery. The impact of this one freely available molecule has been phenomenal—within 10 months this molecule has led to the initiation of proprietary programs, opened up a new area of science, enabled the establishment of a new biotech, and importantly, experts in pharma are trying to convert this target into a new drug for patients.”
Overall, speakers were upbeat about the prospect of epigenetics being able to deliver. Dr. Kouzarides summarized, “We hope to soon see the first potent epigenetic-derived drugs in the clinic. Many of the initial inhibitor molecules such as I-BET and JQ1 may or may not work, but they will establish the principle that epigenetic-derived drugs can be selective and specific.
“Novel druggable targets may be found in other chromatin modification pathways, so in the next few years we might see a shift away from kinase-based drug discovery and toward the epigenetic arena.”

