
October 15, 2014 (Vol. 34, No. 18)
Long-Read Sequencing Can Offer the Most Comprehensive View Yet of Gene Activity
Not too long ago, the life sciences community was still debating whether sequencers would ever overtake microarrays as the preferred means of measuring gene expression. Today, not only have sequencers become the standard workhorse for gene expression studies, but newer sequencing technology has delivered the ability to generate novel expression data even in the most well-characterized cells or organisms. Truly, it is a remarkable time for comprehensive studies of which genes are being transcribed, with the goal of providing functional insight into various biological processes.
The key advantage sequencing holds over microarrays is its ability to deeply survey an entire transcriptome, while microarrays are limited to interrogating known genes using probes designed from a reference genome assembly. As next-generation sequencing became more affordable, scientists were eager to switch to this approach, which became known as RNA sequencing or simply RNA-seq.
Recently, scientists have begun applying long-read sequencing to further advance the field of gene expression, finding that this method can directly sequence full-length transcripts and provide additional insights into gene isoforms. In doing so, this technique has generated a more comprehensive view of full-length, protein-coding gene transcription than other sequencing technologies for the clearest view yet of a transcriptome.
From Short Reads to Long Reads
As a method, RNA-seq has flourished since it was first introduced some seven years ago. Scientists performing RNA-seq studies convert their RNA of interest into cDNA and then sequence it on massively parallel, next-gen sequencers. This generates millions of reads—far more data than it was possible to collect in a single microarray study.
The challenge with most next-gen sequencers, however, is that the reads they produce are quite short (generally maxing out at just a few hundred bases). For gene expression studies, these snippets of information become problematic during assembly, when algorithms have a hard time correctly mapping these reads. This can make isoforms difficult to see, often conflating alternately spliced isoforms into a smaller number of transcripts than they really represent. In some studies, researchers have found that gene isoforms are significantly underrepresented in short-read assemblies.
In a recent review of RNA-seq in Nature Methods, Stanford genetics professor Michael Snyder was quoted as saying, “The way we do RNA-seq now is … you take the transcriptome, you blow it up into pieces and then you try to figure out how they all go back together again. … If you think about it, it’s kind of a crazy way to do things.”
A separate publication in Nature Methods from members of the RNA-seq Genome Annotation Assessment Project (RGASP) presented an evaluation of two dozen protocols in various organisms for inferring transcript information and gene expression level from RNA-seq data. (Such inference is necessary, the authors note, because technical limitations of RNA-seq result in “partial sequence reads of fragmented gene products,” which mandates a shotgun approach to these sequences.)
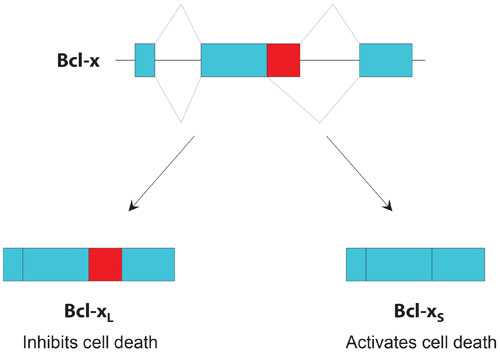
Bcl-x is a classic example of how two gene isoforms can have opposite biological effects, depending on whether a particular exon is retained or spliced out.
One challenge identified through this large benchmarking effort was that transcript identification is quite limited when reads miss exons. “For a significant fraction of transcripts not all exons are identified, ranging from 30% in C. elegans to greater than 60% in H. sapiens,” they write.
Another significant challenge found in the study was reconstruction of isoforms. “These results underscore the difficulty of transcript assembly, which relies on two outcomes: all exons comprising a given transcript must be identified, then connected to form the correct isoform structure,” according to the paper. The researchers determined that automated methods for transcript assembly failed to identify every exon for most transcripts, and that even when all exons were identified, the methods were often unable to pull them together into complete isoforms. “Assembly of complete isoform structures poses a major challenge even when all constituent elements are identified,” the authors report.
The RGASP publication concluded that “unannotated transcript isoforms assembled from RNA-seq data should be interpreted with care, and those critical to an experimental study subjected to independent validation.” Looking to the future, they write, “Ultimately, the evolution of RNA-seq will move toward single-pass determination of intact transcripts. Third-generation instruments will realize that potential and inspire new computing approaches to meet the next wave of innovation in transcriptome analysis.”
One such third-generation technology is Single Molecule, Real-Time (SMRT®) Sequencing from Pacific Biosciences, which generates long reads that have been used by scientists to capture full transcripts from the 5´ end all the way to the 3´ end. In DNA sequencing, this technology generates reads averaging more than 8,000 bases.

The transcriptome is extraordinarily diverse and complex. A myriad of variation can form from individual genes, such as alternative splicing of exons (the portions of the gene that encode for protein), alternative first and last exons, cassette exons where only one or another exon, but not both, are present in a particular mRNA, and many others
Full Transcripts
The ability to perform RNA sequencing with long-read technology is still relatively new, but several pioneering publications describe studies that have utilized the approach. They often report finding critical elements that were previously missed by short-read sequencing. One such study is presented here.
A paper in Proceedings of the National Academy of Sciences from lead author Kin Fai Au and senior author Wing Wong at Stanford University, with several collaborators, used transcriptome sequencing to analyze human embryonic stem cells. The addition of SMRT Sequencing to an existing RNA-seq data set from short-read sequencing helped the scientists characterize more than 13,000 full-length transcript isoforms. More than a third of the isoforms seen in this well-characterized cell line were novel, the scientists reported, noting that long, noncoding RNAs were more likely to be missed by short-read sequence data. The SMRT Sequencing data was useful not just for more comprehensively identifying isoforms, as they found 273 new genes in the data as well. The authors concluded that “gene identification, even in well-characterized human cell lines and tissues, is likely far from complete.”
In addition to having a high-quality genome assembly, it is important to understand all of the gene products, both in terms of their frequency and with regard to the diversity of the different forms that can be created from a particular gene.
Going Forward
With extraordinarily long sequence reads, scientists will be able to learn more about transcriptomes than has ever been possible. Already, studies are turning up new discoveries that simply could never have been detected with short-read sequence data. The ability to accurately identify all transcripts, correctly sort alternately spliced regions, and find important elements such as long, noncoding RNAs will be essential to accelerating our understanding of how gene expression functions.
Jonas Korlach, Ph.D. ([email protected]), is CSO at Pacific Biosciences.

