
September 15, 2008 (Vol. 28, No. 16)
Chemical Labeling Offers Method for Whole Proteome Absolute Quantitation
Proteomics is rapidly progressing with many important applications in protein biomarker discovery and validation. Given the high number of proteins used in the clinic, proteomics is poised to have a massive impact on in vitro diagnostics (IVD), drug development, and rapid assay development.
The high diversity of proteins in any given proteome is reflected by a variety of workflows and study designs intended to achieve qualitative as well as quantitative coverage of the proteins. As all attempts for complete proteome coverage in complex samples have failed, most experienced groups have developed strategies to focus on selected sub-proteomes of specific interest, making quality control and the comparison of proteomics study results impossible.
Over the last few years, the increasing number of proteomics studies has led to thousands of nonvalidated protein biomarker candidates for which little supportive data is available. The data that is published is often then cited as confirmatory in later publications. This is not only detrimental to the anticipated scientific and medical progress but also impacts negatively on the ability of the growing number of sample repositories and biobanks to support biomarker discovery, validation, and development of IVDs and drugs.
This process is currently aggravated by increased access by nonexpert groups to sophisticated MS instrumentation. As a consequence, in the hands of inexperienced groups data generation based on the improved resolving power of mass spectrometers continues to outperform the ability of many users for data interpretation. In addition, the number of samples analyzed in single studies is not increasing to match appropriate statistical methods but rather is stable or even decreasing. As a result, a major impediment to the smooth and swift transition of biomarkers from proteomics research to healthcare is the verification and validation bottleneck.
Reference Materials
Reference materials are widely used for in vitro diagnostics to support the generation of reliable results and their correct use for diagnostic or prognostic purposes. Despite the existence of a collection of standard materials, e.g., for the calibration of IVD devices, no standardized reference materials tailored for proteomics are available today. Regarding this deficiency, efforts are currently under way to remedy that situation. These materials will be used to calibrate instruments and can serve as external references for benchmarking purposes.
Isotope-Dilution Mass Spec
Based on recent developments in quantitative mass spectrometry, biological reference materials have been generated that can be used as internal (quality) controls for proteomics utilizing the characteristics of isotope-dilution mass spectrometry (ID-MS).
ID-MS is recognized as one of the best quantitative technologies in bioanalytics. In the last few years, ID-MS has found widespread application in proteomics. Usually synthetic or recombinant proteins are used as isotopic standards and spiked into samples to serve as internal standards for quantification. Currently standards must be generated one by one by incorporating isotope-labeled amino acids into the peptide sequence by chemical synthesis or by addition to culture media.
Conversely, chemical labeling, attaching small tags that carry isotope differences to the analytes, presents a simpler approach without the need to actually synthesize any peptides. In general, two different methods of chemical labeling are used—isotopic mass labeling and isobaric mass labeling.
Isotopic mass labeling leads to a distinct mass difference of structurally identical reference and target analyte. The ratio between the heavy and light analyte are used for quantification. Isobaric mass labeling leads to isobaric analytes that can be differentiated by isotopic mass reporters released from the analyte during tandem mass spec. Again, the ratio between different mass reporter intensities is used for quantification
For both modes, absolute quantification is achieved by spiking in the reference in known amounts. Otherwise, relative quantification values are obtained.
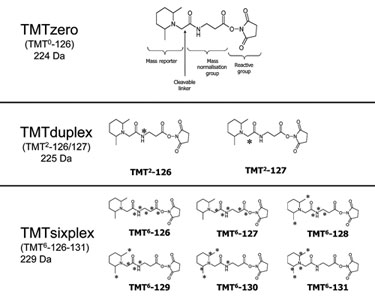
The Tandem Mass Tag® (TMT®) core structure invented by Proteome Sciences allows for the generation of different sets of isotopic and isobaric mass tags. TMTduplex tags incorporate one 13C each, giving an isobaric set of two tags (Figure). TMTsixplex tags incorporate four 13C and one 15N, giving an isobaric set of six tags. Combining TMTzero with either TMTduplex or TMTsixplex gives a set of isotopic tags, as would a combination of TMTduplex with TMTsixplex.
The principle behind chemical labeling is that the quantitative ratio between individual proteins from different samples remains constant after labeled samples are mixed. This conservation is the key for subsequent multistep separation and analysis, providing much better control, precision, and accuracy.
Mixing of labeled samples should be performed as early as possible during any workflow, and one of the samples needs to be a common reference sample
The most commonly applied labeling method uses the reaction between protein amine functions and succinimide esters, resulting in labeling of >98% of amine functionalities. As several amine functionalities (amino-terminal amino acids and epsilon-amine functions of lysines) are present in almost all (human) proteins, virtually the entire proteome is accessible to mass spectrometric quantification with this technology.
Figure
Biological Reference Materials
In contrast to synthetic reference materials, which are limited to a few analytes, chemical labeling allows for the labeling of the entire proteome, thus creating a full biological reference. Using chemical-labeling materials as control samples will make it possible for scientists to have quality control and improved quantification and to compare their results more easily, notwithstanding differences in workflow and instrumentation. Since these standard materials can be used by multiple labs, cross-study and cross-lab comparisons are now possible.
Targeted Assays for Protein Biomarkers
To allow for the specific measurement of a protein biomarker by MS, proteotypic peptides, which are generated by (tryptic) digestion and are unique to the candidate biomarker, are used. The gold standard methods in both their sensitivity and specificity are selected reaction monitoring (SRM) and multiple reaction monitoring (MRM) on triple quadrupole mass spectrometers.
The target analyte is quantified by simultaneous measurement of a spiked, isotope-labeled reference analyte of identical chemical composition. By utilizing two isotopic tags, multiple peptide pairs from reference and sample are generated that exhibit identical elution and fragmentation properties, while also being isotopically distinct. This allows for the rapid transition of discovery results into highly specific and sensitive assays.
When an identical aliquot of the labeled biological reference is spiked into individually labeled study samples, all biomarker candidates from a discovery study can be transferred into a reference-controlled SRM/MRM analysis on a triple quadrupole mass spectrometer within days to weeks.
Utility of Labeled Biological Reference Materials for Proteomics
Peter Schulz-Knappe is CSO at Proteome Sciences. Web: www.proteomics.com. Email: [email protected]. TMT and Tandem Mass Tags are registered trademarks of Proteome Sciences. Tandem Mass Tags are available through Pierce, a Thermo Fisher Scientific company.

