
January 15, 2017 (Vol. 37, No. 2)
With the Right Conditioning Raw Samples May Become More Manageable
Proteomics lags behind genomics and transcriptomics, endeavors that have been accelerated by next-generation sequencing. Although proteomics isn’t exactly stuck—it is chugging along thanks to mass spectrometry (MS)—it sometimes spins its wheels because the world of proteins is particularly slippery.
Proteins may reflect splicing events or post-translational modifications. Proteins occur in concentrations that cover an immense dynamic range. And proteins form complexes and participate in protein networks that are variable over time and space.
If proteomic analysis is to overcome these challenges, it will need more traction at every stage, starting with sample preparation. Improvements at this stage may be especially important, since lapses here may be beyond correction at subsequent stages, which include the generation and detection of target fragments by MS, as well as the analysis of MS data by computational systems.
Current sample-preparation techniques are not always satisfactory. Expedients include enrichment, a means of bringing low-abundance proteins to light, and depletion, a means of looking past high-abundance proteins. Yet these techniques may inadvertently exclude or degrade proteins of interest. Worse, they may fail to capture contextual information or generate artifacts, which may confound attempts at analysis later.
Alternatives to the current expedients are being developed. They include labeling, separation, and extraction methods that can preserve contextual information. For example, there are new methods that could keep protein-protein complexes from falling apart before they are detected. Also in development are approaches that could improve our ability to see patterns of protein localization within the cell.
Wider perspectives can be acquired by tracking the dynamic cell-to-cell variations in proteins. And should a tighter focus be desired, there are techniques that can peer into niches that harbor elusive forms such as small peptides or post-translationally modified proteins.
Advances in understanding ribosome structure and function are shaping new directions in proteomic discovery. The 3D image is a model of the human ribosome. RNA of the large subunit is shown in purple, and RNA of the small subunit in blue; tRNA is shown in white, and proteins in rainbow. [NIH 3D Print Exchange, 3dprint.nih.gov]
Post-Translational Analysis
“Challenges in studying protein post-translational modifications extend to both identification and quantitation,” says Yue Chen, Ph.D., assistant professor of biochemistry, molecular biology, and biophysics at the University of Minnesota. The identification and characterization of protein modifications is particularly challenging for low-abundance proteins, which are often detected only after enrichment.
Enrichment technologies have been developed, and several of them have proven to be useful. None of them, however, is so useful that it can serve in every instance. Typically, each technology works well only for selected post-translational modification types. Therefore, identification of low-abundance protein modifications still depends on the availability of specific enrichment strategies.
Quantitative proteomics strategies such as SILAC and iTRAQ have been widely applied to study the dynamics of proteome. However, a critical limitation to quantify post-translational modifications is that these strategies only quantify changes in their relative abundance.
“It is very difficult to measure absolute stoichiometry of protein modifications in mass spectrometry experiments,” notes Dr. Chen. “The technology for this is not yet mature.”
While relative quantitation provides valuable information, it lacks a layer of data that can only be extracted from the absolute quantitation of the respective changes. For any post-translational modification, the same degree of relative change may reflect very different changes in its absolute abundance, and can therefore differ in terms of physiological and functional relevance.
In a recent study, Dr. Chen and colleagues developed a quantitative proteomics strategy to stoichiometrically identify site-specific lysine acetylation in mammalian cells. This approach used the stable heavy isotope-based labeling of the unmodified lysine residues with acetyl groups, followed by deep proteomic profiling and the use of a novel in-house-developed software program for quantitative analysis for stoichiometry determination.
A key advantage of this approach was that it allowed not only the relative, but also the absolute stoichiometry to be interrogated in a global manner. “This approach does not rely on enrichment techniques,” elaborates Dr. Chen. “Accordingly, it provides an unbiased view of the presence and stoichiometry of lysine acetylation at a proteome-wide level.”
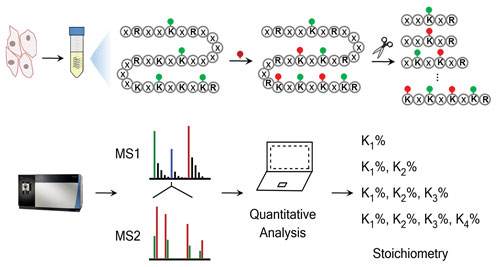
A chemical proteomics workflow for global and site-specific measurement of lysine acetylation stoichiometry. [University of Minnesota]
Protein-Protein Interaction Analysis
Capturing protein-protein interactions is one of the most widely used experimental approaches, critical for virtually all areas of life sciences. However, characterizing the interaction between proteins is challenging.
“In many instances, it is impossible to develop separation conditions that allow the detection of an intact complex,” says Robert Kennedy, Ph.D., professor and chair of the chemistry department at the University of Michigan. “Many factors may contribute to detection difficulties. To overcome these difficulties in the analysis of peptides and proteins, we are using miniaturized separation methods.”
An ongoing effort in Dr. Kennedy’s lab has been the use of miniaturized techniques to measure noncovalent protein-protein interactions. Miniaturization provides two advantages in this context. “When the electrophoresis column is miniaturized, it is possible to apply higher voltages and perform faster separation,” says Dr. Kennedy.
Therefore, weakly interacting proteins, which form complexes with a shorter lifetime, can be separated before they dissociate, allowing the detection of the complex. Additionally, this approach uses much less sample. “We want to use this approach to develop rapid assays that can be used for screening interventions,” says Dr. Kennedy. “Ultimately, we want to identify from a library those molecules that modulate an interaction.”
In a recent study, Dr. Kennedy and colleagues described a new way to use capillary electrophoresis in protein-protein interaction analysis. The new approach, protein cross-linking capillary electrophoresis (PXCE), can solve a difficult problem—preserving protein complexes during separation.
When two interacting proteins are mixed, the noncovalent complex are separated from the free protein to quantitate the bound-to-free protein ratios. Somehow, separation conditions must prevent protein adsorption to the capillary and maintain protein interactions. In the new method, the proteins are cross-linked before separation.
“Rather than capturing a noncovalent complex, we can freeze it by rapidly crosslinking it with reagents such as formaldehyde, and then separate it,” explains Dr. Kennedy. “Basically we dissociate the interaction step from the separation step.”
Dr. Kennedy and colleagues recently reported that they used PXCE to evaluate antibody–antigen interaction and heterodimer and homodimer heat shock protein complexes. The investigators asserted that PXCE “gives good quantitative results” and “allows rapid method development for quantitative analysis of protein-protein interactions.”
A critical aspect of these assays is the availability of compounds that react fast enough to capture even weak interactions between proteins. “Fast reagents and rapid mixing, performed in a way that allows us to conduct high-throughput experiments, would move the field forward,” insists Dr. Kennedy.
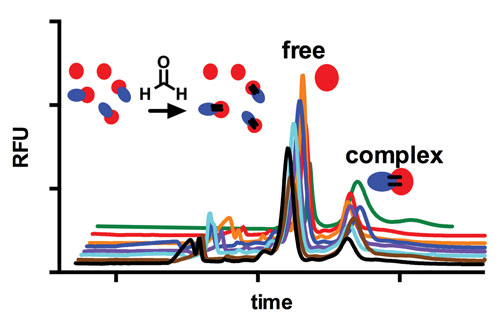
Schematic and electropherograms of protein cross-linking capillary electrophoresis experiment for analysis of protein-protein interactions. [University of Michigan]
Cell-Signaling Analysis
“Mass spectrometry has now matured as a technology to the point that enables the highly quantitative, accurate, and increasingly near-comprehensive analysis of biochemical signaling in cells,” says Alex Kentsis, M.D., Ph.D., assistant professor and group leader at the Memorial Sloan Kettering Cancer Center.
One of the challenges in mass spectrometry revolves around identifying the most effective strategy to extract the cellular proteome or its subsets. Various experimental approaches show different biases when they are used to capture proteins localized in specific cellular compartments.
To identify the most effective experimental approaches for native proteome extraction, Dr. Kentsis and colleagues performed a systematic analysis of cellular lysis and protein extraction methods. Then the investigators used the information to combine a high-efficiency mechanical disruption system with an optimized detergent system. These systems were combined to maximize the extraction and minimize the degradation of native proteins.
Dr. Kentsis’ laboratory focuses on developing new methods to quantitatively analyze cellular signaling and help interrogate a variety of biological questions. “We would like to convert these methods into clinical tests,” notes Dr. Kentsis. He adds that the conversion “requires additional analytical performance features that would ensure the accuracy and the reproducibility that are needed for clinical diagnosis.”
High-resolution MS is finding application in the clinic more slowly than it is finding application in the research laboratory, for example, in studies of bacterial lysates and tissue extracts. “Partly this is due to the relatively specialized nature of the instrumentation,” explains Dr. Kentsis. “But highly accessible and robust techniques are increasingly being developed.”
Peptidomic Analysis
“The instruments have been great in looking at the top 10% of the proteins or peptides,” says Lloyd D. Fricker, Ph.D., professor of molecular pharmacology at Albert Einstein College of Medicine. “But going deeper into the proteome or peptidome has been more difficult.”
Research in Dr. Fricker’s group is focusing on peptidomics, an area within proteomics that involves peptide profiling and uses experimental approaches similar to proteomics. Whereas proteomic analysis is preceded by the digestion of proteins into peptides, peptidomics skips this initial step because it analyses peptides that are already present in biological samples.
In a recent study that used a quantitative peptidomic technique, Dr. Fricker and colleagues revealed that budding yeast cells contain peptides that are derived from intracellular proteins. The investigators showed that these peptides are very unlikely to be artifacts of the cellular extraction process. Most of the peptides that were detected originated from proteins that were among the top quintile in terms of their cellular abundance.
The yeast and the human peptidome captured in this analysis were similar in terms of average peptide size and amino acid composition. A quantitative comparison between the budding yeast and the mammalian peptidome revealed that many of the peptides were involved in protein synthesis, protein folding, and cellular metabolism. The comparison also pointed to a high degree of conservation between the yeast and the human peptidomes.
Dr. Fricker’s group has also found that the relationship between the abundance and the importance of proteins is not as obvious as might be imagined. That the most abundant proteins should fulfill important cellular roles occasions little surprise. However, many of the less abundant proteins have been found to perform critical functions, too.
“Some of the peptides that are present in a neuronal subpopulation can have very specific functions and behaviors,” Dr. Fricker points out. “In the mouse brain, this subpopulation accounts for just 10,000 neurons, which is not a lot, considering the mouse brain has billions of neurons.”
Although this observation is intriguing, it is also frustrating. Digging deeper poses the difficult or nearly impossible task of detecting peptides that originate from a relatively small group of neurons, a group that is part of a larger sample, a large brain region or even the entire brain.
A critical limitation in proteomic and peptidomic analysis is sensitivity, a parameter that is intimately shaped by the cellular abundance of a protein, which can vary broadly in the same cell, depending on the protein. Proteins can range from a few copies to one million copies per cell. “At this time, MS cannot detect peptides that are present at very low levels because many copies of a molecule are needed in order to produce a signal,” explains Dr. Fricker.
An area that is open for improvement, and promises to shape sensitivity, is the development of MS equipment that cycles faster. In electrospray ionization mass spectrometry, peptides are separated through a column and eluted. Subsequently, the instrument cycles through up to 10–20 peptides at one time and fragments them, before picking up another group of peptides. “The most abundant peptides are detected first, but as the cycle time improves, the machine can detect less abundant peptides,” informs Dr. Fricker.
Another much-needed development, critical for both proteomics and peptidomics, involves improvements in liquid chromatography. “There will be new applications in which existing technologies can be applied to break down a sample into subsamples,” predicts Dr. Fricker. This capability, he continued, could “subject a greater diversity of the material to analysis.”
Biomarker Identification
“The idea behind our work is to help provide a better statistical and biological manner to detect prostate cancer and determine at what stage it may exist,” says Paul A. Townsend, Ph.D., professor of molecular cell biology and associate dean at the University of Manchester.
Prostate cancer is among the most common malignancies in males worldwide, and survivors still face high mortality rates despite the availability of screening. Prostate-specific antigen (PSA) has been used as a screening test for prostate cancer, but its level in blood can also increase in other conditions, including inflammation. Furthermore, overdiagnosis emerges as a critical problem, due to the treatment-related adverse effects that have long been described. These therapeutic challenges point toward the need to develop better diagnostic tools.
“Generally, one cannot identify a single molecule that correlates with disease,” observes Dr. Townsend. “Instead, there are always multiple proteins that, together, in the way that they are upregulated or downregulated, give a profile that correlates with disease.”
A major effort in Dr. Townsend’s group has been the development of better screening tools by identifying molecular fingerprints or molecular targets that, when used together, as a panel, are informative about illness or health. A critical prerequisite for a diagnostic test is its noninvasive nature, which increases its acceptance by target populations. “Collecting a blood sample is a lot easier and less stressful than an examination or a biopsy,” notes Dr. Townsend.
The biomarker that Dr. Townsend and colleagues developed relies on the use of serum, which is easy to obtain. Using blood samples for developing biomarkers presents at least one other advantage. “If the body perceives the presence of the illness in any way, there is a systemic response, and the best medium to assess that has to be the blood,” Dr. Townsend points out.
Serum and the blood, however, are extremely complicated biological fluids. They present several layers of complexity, which opens challenges for their use in proteomics experiments. By mass, over half of the serum proteins is albumin. The abundance of this carrier protein may mask the less abundant proteins in the serum and make their detection and analysis challenging. Many proteomic strategies deplete the highly abundant proteins in the serum, such as albumin and immunoglobulins, but inadvertently remove some less abundant proteins that otherwise could provide valuable data.
The strategy used by Dr. Townsend and colleagues, iTRAQ 3D-LC-MS, did not deplete the serum sample, but instead it fractionated the various classes of proteins into groups based on their molecular size. “Therefore, in theory, this approach would never miss a proteomic signature,” asserts Dr. Townsend. Although this new strategy is more expensive and time-consuming than conventional techniques, it provides more signal. “It provides the opportunity to look at everything in situ, rather than at a selected package of the information,” emphasizes Dr. Townsend.
Dr. Townsend and colleagues, especially Spiros Garbis, Ph.D., associate professor, head of Proteomics Unit for Cancer Sciences, Centre for Proteomics Research, Institute for Life Sciences, University of Southampton, built on previous research to develop a whole-serum analysis approach. The investigators used it to identify biomarkers informative about prostate cancer progression.
The whole-serum strategy identified several putative targets out of a large collection of proteins initially identified through conventional experimental strategies such as ELISA. One thousand proteins were winnowed down to a group of six proteins that form an interactome and represent a promising framework to interrogate their utility as biomarkers.
This experimental approach provides a proof of principle to develop better biomarkers for other conditions. “This tool can be applied to cancer or any disease—anything that impacts health and can be monitored at the level of the blood,” insists Dr. Townsend.
Mass Spectrometry for Clinical Proteome Analysis
Proteomics is a rapidly growing field of molecular biology that involves the systematic, large-scale, high-throughput analysis of protein expression in cells and organisms using mass spectrometry.
By detecting, quantifying, and characterizing the full range of proteins in a given sample, key biomolecular differences between normal biology and disease states can be identified. This information can help researchers understand how diseases act on the body, and may potentially be used for disease diagnosis, particularly in patients that present otherwise normal physiology, such as those with cancer.
Precise Measurements
These experiments can involve analyzing anywhere in the region of 50 to 5,000 proteins or fragments in a single sample. Highly sensitive and precise mass spectrometry measurements are therefore necessary to distinguish between the often large range of proteins of varying cellular concentration and degree of modification by post-translational events.
“To overcome this challenge, researchers are utilizing the ultra-high sensitivity, accuracy and resolving power offered by the latest quadrupole-Orbitrap instruments,” said Andreas Huhmer, Ph.D., director, proteomics and metabolomics marketing, Thermo Fisher Scientific. “In one study, researchers set out to identify trypsin protein cleavage sites in sarcomere muscle tissue in order to better understand how diseases affect muscle contraction and cause muscle atrophy.
“After partially digesting the muscle motor protein myosin (responsible for contraction) into meromyosin sub-units, the team used a proteomics approach to perform a functional assay on the digestion fragments to identify where the protein was cleaved and reveal trypsin-resistant protein domains.”
Whereas gel electrophoresis could not resolve trace digestion components from more concentrated fragments, the ultra-high measurement resolution and accuracy of the quadrupole-Orbitrap instrument allowed low-intensity fragments to be detected in the presence of components 1,000 times more concentrated, according to Dr. Huhmer.
Proteomic Analysis Used to Link RA and Gum Disease
The idea that gum disease can be a driver of more detrimental systemic disorders has been kicked around the immunological community for a number of years; however, the molecular evidence linking the ideas together has been slow to accumulate. Yet now, investigators at Johns Hopkins University report they have new evidence that a bacterium, known to cause chronic inflammatory gum infections, also triggers the inflammatory autoimmune response characteristic of chronic, joint-destroying rheumatoid arthritis (RA).
“This is like putting together the last few pieces of a complicated jigsaw puzzle that has been worked on for many years,” remarked senior study investigator Felipe Andrade, M.D., Ph.D., associate professor of medicine at the Johns Hopkins University School of Medicine.
The findings from this study recently appeared in Science Translational Medicine, in an article entitled “Aggregatibacter actinomycetemcomitans-Induced Hypercitrullination Links Periodontal Infection to Autoimmunity in Rheumatoid Arthritis.” It describes a common denominator the Hopkins team identified in periodontal disease and many people with RA—the bacteria A. actinomycetemcomitans (Aa). Infection with Aa appears to induce the production of citrullinated proteins, which are suspected of activating the immune system and driving the cascade of events leading to RA.
“This research may be the closest we’ve come to uncovering the root cause of RA,” noted lead study investigator Maximilian Konig, M.D., a former Johns Hopkins University School of Medicine fellow now at Massachusetts General Hospital.
The clinical association between periodontal disease and RA was first noticed in the early 1900s, and over time, researchers have suspected that both diseases may be triggered by a common factor. In recent years, studies have focused on a bacterium known as Porphyromonas gingivalis, found in patients with gum disease. However, while major efforts are currently ongoing to demonstrate that this bacterium causes RA by inducing citrullinated proteins, all attempts by this research team have failed to corroborate such a link. Dr. Andrade and his colleagues have persisted on finding alternative bacterial drivers.
“We used mass spectrometry to define the microbial composition and antigenic repertoire of gingival crevicular fluid in patients with periodontal disease and healthy controls,” the authors wrote. “Periodontitis was characterized by the presence of citrullinated autoantigens that are primary immune targets in RA. The citrullinome in periodontitis mirrored patterns of hypercitrullination observed in the rheumatoid joint, implicating this mucosal site in RA pathogenesis.”
Citrullination happens naturally in everyone as a way to regulate the function of proteins. However, in people with RA, this process becomes overactive, resulting in the abnormal accumulation of citrullinated proteins.
The Hopkins team found that Aa was the only pathogen able to induce hypercitrullination in neutrophils, which are highly enriched with the peptidylarginine deiminase (PAD) enzymes required for citrullination. Neutrophils are the most abundant inflammatory cells found in the joints and the gums of patients with RA and periodontal disease.
Moreover, the investigators found that Aa initiates hypercitrullination through the bacterial secretion of a toxin—leukotoxin A (LtxA)—as a self-defense strategy to kill host immune cells. The toxin creates holes on the surface of neutrophils, allowing a flux of high amounts of calcium into the cell where concentrations are normally kept low. Since the PAD enzymes are activated by calcium, the increased exposure overactivates these enzymes, generating hypercitrullination.
Gathering 196 samples from a study of RA patients, the researchers found that almost half of the patients—92 out of 196—had evidence of infection by Aa. These data were similar to those for patients with periodontal disease, but quite different from those in healthy controls. More strikingly, exposure to Aa was a major determinant in the production of antibodies to citrullinated proteins in patients with genetic susceptibility to RA. The scientists warned against overinterpreting the results, as more than 50% of the study participants who had RA had no evidence of infection with Aa. Other bacteria, in the gut, lung, or elsewhere, could be using a similar mechanism to induce hypercitrullination.

