
August 1, 2010 (Vol. 30, No. 14)
Single-Cell Technique Embraced for What It Can Reveal about Molecular Mechanisms
As technology continues to evolve, the potential of gene-expression profiling continues to grow. The field is now expanding to focus on single cells, which many believe have unlimited potential for molecular biologists. A number of companies and academicians presented their latest single-cell related advances at the recent BioEPS “qPCR” meeting in Vienna.
Fluidigm’s Biomark™ System has a relatively new dynamic array biochip (96.96 array) that tests 96 cells against 96 genes in one experiment. “One can assay single cells with a classic PCR microtiter plate, but it’s difficult to run more than an experiment or two off the material from a single cell,” explained Howard High, company spokesman.
“Fluidigm’s platform can run 96 experiments off the contents of a single cell, as well as run several plates. We’ve had customers run up to 1,000 experiments from one cell.” The array assembles cDNA material from individual cells with reagents to create individual qPCR reactions.
This provides significant savings, added Ken Livak, Ph.D., senior scientific fellow. Using standard 384-well plates, it would take about 8 days for a single instrument to run 96 samples against 96 assays. A single run with the chip provides the same throughput as 24 384-well plates, but in only 4 hours, while using 1/200th the amount of reagent, Dr. Livak said. The application to single-cell analysis provides insight into developmental signals such as stem cell differentiation. “When you look at the average gene expression from a large number of cells, you can obscure signal. You’re looking at an average of 10,000 cells, so it doesn’t really tell you what’s occurring among individual cells.”
According to Dr. Livak, analysis of single cells is complex because there are fluctuations in gene expression within single cells. This makes it important to look at enough individual cells to understand the temporal fluctuation and variation that is occurring.
Current applications of the Fluidigm technology include analysis of stem cells and cancer, both of which require single-cell gene-expression profiling to understand, Dr. Livak noted. In addition, the platform is being used in immunology, T-cell studies, and neurological research. “This may be useful for individualized medicine for a specific tumor and to assess the stage of cancer and how aggressive it has to be treated.”
“As people dive into the single-cell arena, the opportunities are limitless. There are so many questions to be answered and so many people trying to figure out the right questions. This is exploding on a global scale,” summarized High.

Fluidigm reports that its 96.96 Dynamic Array™ integrated fluidic circuit can deliver 9,216 individual real-time PCR reactions with 192 pipetting steps.
miRNA Expression
Researchers at the University of British Columbia’s Center for HT Biology presented a large-scale profile of miRNA expression across purified hematopoetic populations using Fludigm’s dynamic array. The study included 27 blood cell populations, of which 8 are rare. The arrays tested 300 miRNAs and saved substantial time by cycling 2,300 samples per run.
The basis of the study was to examine how miRNA is expressed in each population and how it varies across different lineages—what changes as cells are restricted in their potential toward a final cell type. “That’s an important biological question in hematopoesis. Prior to this study, no one had an idea of the miRNA profile across these cells,” stated Carl Hansen, Ph.D., assistant professor, applied physics. “I think this is the first big picture of miRNA regulation across hematopoetic development.”
Study results showed that there are signatures of miRNA that are more similar to the primitive types than the direct descendants of the primitive cells. Another observation was that there are no miRNA specific to a given cell type. So it seems unlikely that one miRNA could turn an entire genetic program on or off. However, Dr. Hansen added, there are patterns that indicate what a cell type is and how it’s related.
Another part of the study looked at miRNA expression in single cells. There is low variation in the miRNA; according to Dr. Hansen, within their measurement error they were unable to differentiate single cells from each other. This indicates that the miRNA is tightly regulated at the single-cell level, which suggests that profiling miRNA would be a good way to estimate heterogeneity of a population, as well as to identify rare cell types within that population.
Dr. Hansen emphasized that when reviewing the final, raw miRNA data with real-time PCR, it’s important to normalize the biases. His group did this by taking a synthetic pool of all the miRNA being tested and doing a dilution series. This provided a better idea of the differences between cell types.
Potential clinical applications for miRNA profiling include cancer prognosis and therapy responsiveness. “All this research is intended to better understand these pathways and develop better treatment and diagnosis.”
miRNA PCR System
Some of the main challenges in working with miRNAs include short sequences, close relatation between family members, and high variability of the C-G content—all of which cause different melting temperatures and make it difficult to get specific assays without a lot of background noise.
These issues are addressed with Exiqon’s miRCURY LNA Universal RT microRNA PCR. The combination of a universal RT reaction with the company’s LNA™-enhanced PCR primers reportedly results in high sensitivity and specificity.
There are two new ready-to-use panels that contain pre-aliquoted miRNA PCR primer sets in 384-well PCR plates—the version 2.0 human panels enable profiling of up to 742 human miRNAs using only 40 ng total RNA, according to the company.
LNA™ (locked nucleic acid) is a high-affinity nucleotide analog with a different sugar backbone structure than RNA and DNA, yet it is reportedly easily incorporated into oligonucleotides using standard synthesis procedures. Correct incorporation of LNA in PCR primers results in sensistive and specific PCR assays, explained Peter Mouritzen, director, life science product development. “LNA-enhanced PCR primers will discriminate against a single nucleotide mismatch, and thereby, provide the ability to discriminate closely related miRNAs, as well as between mature and precursor microRNAs.”
It also offers high sensitivity and low background for accurate measurement of miRNA expressed at low miRNA levels. In addition, studies have shown that LNA-enhanced primers enable working with difficult sample types like serum plasma and FFPE sections. “We have some pilot studies that suggest that even single-cell analysis is possible,” added Ditte Andreasen, Ph.D., senior scientist.
A large discovery project on serum plasma showed good results—detecting a large number of microRNAs, in a reproducible manner. “The cell-free serum and plasma samples are the most challenging sample types because the total miRNA level is low and there is such a high risk of carrying inhibitors over,” said Dr. Andreasen.
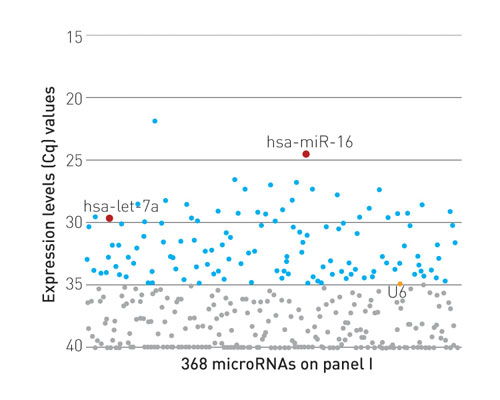
miRNA expression profiling of 368 miRNAs using total RNA purified from 35 µL serum [Exiqon]
RT-qPCR
Researchers at the Lundberg Lab for Cancer Research at Gothenburg University reported that they have optimized the steps in RT-qPCR for more accurate single-cell gene-expression profiling. Anders Stahlberg, Ph.D., investigator, said that it’s important to understand single-cell biology and due to the small number of transcripts being measured, it’s often difficult to separate technical variation from biologically relevant cell-to-cell variation. His group combined various existing methods and focused on enhancing steps in the process.
“RT-qPCR has high sensitivity and reproducibility but hasn’t become common practice for single-cell gene-expression profiling. We used the best of all the steps and optimized them,” Dr. Stahlberg said. Cell collection was done via glass capillaries with a wide tip (~10 µm) to allow passage of intact cells, ensuring collection of all mRNA.
A control for contamination of the buffer surrounding the cells was analyzed with the single-cell samples. Cells must be lysed and all available mRNA accessible to the enzyme. As a control for the workflow, Dr. Stahlberg said, an RNA spike can be added to the lysis buffer, and it should be unique compared to the transcriptome of the single cells being analyzed.
For the reverse transcription step, it’s important to use an optimized gene-specific primer identical to the reverse primer, as well as to test different reverse-transcriptases because efficiency varies greatly. Since the number of genes analyzed from a single cell is limited by the number of transcripts, Dr. Stahlberg said his research shows that more than 20 target molecules are needed for accurate quantification.
Data analysis can be difficult due to the variation in mRNA levels among cells. “You need to normalize to compensate for variation between samples.” This is to distinguish between experimental and biological variability. “People working from more cell population measurements think you should normalize single-cell data against a reference gene, but this is wrong. It’s important to know the principals of single-cell gene-expression profiling and how to standardize it and how to look at the data.”

