
November 1, 2010 (Vol. 30, No. 19)
Cancer and Metabolic Diseases Represent Promising Applications
Histone deacetylases (HDACs) are emerging as exciting new targets for drug development, particularly for cancer and metabolic diseases. Because they exert control over gene transcription and cell-cycle progression, they represent a novel approach for epigenetic therapeutic intervention.
The first such agent was approved by the FDA in 2006 (suberoylanilide hydroxamic acid or SAHA, vorinostat) for the treatment of advanced cutaneous T-cell lymphoma. The rapid approval of SAHA helped accelerate the study of this innovative area. Currently, only one other HDAC is approved in the same indication, although a number of other inhibitors are now in development.
There are many proposed consequences of HDAC inhibition that range from induction of apoptosis, inhibition of DNA repair, upregulation of tumor suppressors, and others. This is further complicated because the HDAC family consists of at least 18 members divided phylogenetically into four classes.
CHI’s conference “HDAC Inhibitors” will showcase updates on new therapeutics and emerging methodologies, as well as discussing where the field is headed.
“There are already some links identified between the presence of specific isoforms of HDAC enzymes and specific forms of cancer,” notes Bernd Hentsch, Ph.D., chief development officer at 4SC. “For example, selected HDAC enzymes are frequently found to be overexpressed in specific types of cancer as compared to normal tissue, such as the HDAC1 protein in hepatocellular carcinoma (HCC) or HDAC2 in colorectal tumors.
“In fact, this overexpression, as exemplified in the case of HCC and HDAC1, can be directly linked to a poorer prognosis. Even though this would imply that targeting such individual HDAC enzymes could be the way to go, the development of an HDAC inhibitor with a wider spectrum could provide broader applicability or even stronger clinical activity.”
The race to identify the most promising approach is still being run, notes Dr. Hentsch. “The field is currently trying to understand whether it would be more advantageous to target individual HDAC proteins or pursue a broader (pan) inhibitory approach. It is clear that if one strikes more HDAC enzymes at a time, such a drug may cover more malignancies or may even offer a more efficacious therapy. On the other hand, the fine-tuned balance between HDAC inhibitor selectivity and toxicity is still unclear at present.”
The company is developing its own lead HDAC inhibitory product, resminostat. “This is a novel pan-HDAC inhibitor undergoing Phase II evaluation as an oral mono or combination therapy for treating advanced HCC and relapsed or refractory Hodgkin lymphoma,” Dr. Henstch says. “We are also due to commence a Phase II trial to investigate resminostat as a second-line treatment for KRAS-mutated colorectal carcinoma patients in combination with standard chemotherapy. To broaden our HDAC portfolio, we are advancing a selective HDAC into a Phase I this year.”
Selective HDAC Inhibition
Although many HDAC inhibitors target cancer, the central nervous system (CNS) represents another lucrative therapeutic domain, notes James Rusche, Ph.D., senior vp, R&D, Repligen. “For example, there are a number of CNS genetic diseases caused by a repetition of a DNA triplet sequence. In Friedreich ataxia (FRDA), a repetitive triplet repeat falls within the first intron of the frataxin gene. This causes hypoacetylation and ultimately blocks normal transcription so that the protein produced is fully functional, there is just not enough of it.”
HDAC inhibitors might provide a new therapeutic strategy for treating FRDA, according to Dr. Rusche. “Research at Scripps showed that one class of HDAC inhibitors could increase the mutant frataxin gene expression. While current HDAC inhibitors for oncology may not work due to toxicity and tissue distribution problems, we felt that we could find better molecules more suited to treating Friedreich ataxia.”
Thus, the company needed to find a more selective HDAC inhibitor that could distribute to brain tissue. “Our preclinical candidate is potent, selective, and possesses the pharmacology needed for an orally active, brain-penetrant drug,” Dr. Rusche reports. “This compound, which is selective for HDAC3, increases expression of the frataxin gene in brain tissue when tested in transgenic mouse models of FRDA at concentrations without side effects. We are now gearing up for clinical trials.”
Other CNS applications that modify histone acetylation will likely follow. “We can envision applications such as Huntington disease, spinal muscular atrophy, and memory modulation. This area of modulating memory is just beginning, but animal studies are providing results that encourage further development in this area.”
Although CNS drug development can be challenging, future HDAC therapeutics are on the horizon. “Most compounds do not get into the brain. The second-generation HDAC inhibitors will likely be much more selective with lowered toxicity and improved delivery to the target tissue. Diseases of the CNS are one important example of how next-generation HDAC inhibitors will provide a much broader opportunity for therapeutics development.
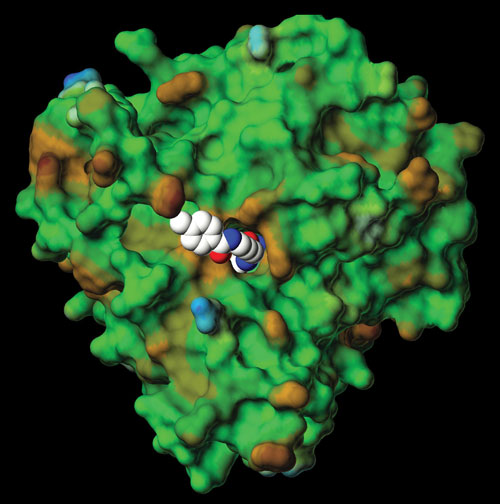
A model of RG2833, Repligen’s lead compound for Friedreich ataxia, docked to the HDAC3 enzyme.
Targeting Approach
Epigenetic agents like HDAC inhibitors have a low therapeutic window. One way to overcome such issues is to utilize a cell-targeting approach for delivery, notes Alan Drummond, Ph.D., CSO and founder at Chroma Therapeutics. “Since HDACs work intracellularly, the main challenge is to limit the number of cells that are exposed to the drug. We have developed a method for intracellular delivery specifically to monocytes and macrophages, which are key protagonists in inflammatory conditions.”
The company’s esterase sensitive motif (ESM) technology involves the attachment of a chemical group onto an HDAC inhibitor. Once inside the monocyte or macrophage, it is cleaved by a specific carboxylesterase to give rise to an active charged species that cannot readily migrate out of the cell. The intracellular accumulation of the pharmacologically active acid leads to enhanced potency and duration of effect, but only in cells expressing this specific enzyme.
“Tolerability is always the potential Achilles heel with epigenetic agents,” according to Dr. Drummond. “We found a direct cell-targeting approach that appears to direct the epigenetic effect to a subset of cells that were important in pathology leading to reduce systemic toxicity. The physiochemical properties of the acid product determine longevity of effect and tolerability, not organs such as the liver. We have now shown up to 1,000-fold selective delivery to human monocytes in blood.
“This technology is an example of how to specifically target drugs such as HDACs and non-HDAC intracellular targets to macrophages/monocytes,” Dr. Drummond reports. “Additionally, such targeting also has applications for treating monocytic tumors, for example in acute myelogenous leukemia subtypes. Finally, new research has revealed that macrophages can play a major role in tumor progression and metastasis. We are currently studying the potential of this technology to target such cells in order to reverse this effect.”
Interrogating Activity
Discovery and characterization of new HDAC inhibitors requires sensitive and accurate high-throughput functional assays. According to Andrew Niles, senior research scientist at Promega, this has been a challenge.
“We evaluated current commercial kits that utilize fluorescence readouts and found they required relatively large amounts of deacetylase enzyme and were difficult to miniaturize into high-density formats. Our approach was to develop a one-step, enzymatically coupled, bioluminescent assay reaction. This system employs an optimized acetylated peptide substrate as an indicator of deacetylase activity, with a proteolytic developer reagent and Ultra-Glo™ luciferase chemistry.”
Niles says the assay is a simple add, mix, and measure format that typically only takes about 15 minutes to achieve a steady-state luminescent signal proportional to deacetylase activity. “The large signal windows and glow-type luminescence are what facilitates rapid high-throughput screening. In contrast, fluorescence methods require multiple steps, take up to two hours, and have less sensitivity.”
Because HDACs consist of four families of isoforms, it is important that functional assays accurately interrogate the various HDAC classes. “Class III HDAC enzymes (sirtuins) utilize a NAD+ dependent deacetylase mechanism, whereas Class I/II enzymes operate in a zinc-dependent manner.”
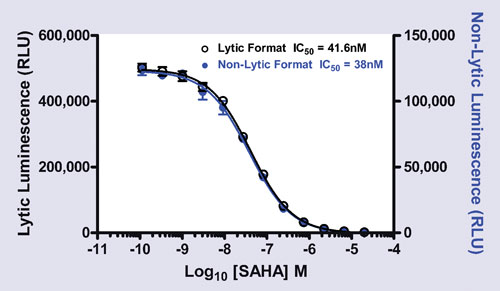
Promega reports that its HDAC-Glo I/II can be used to interrogate deacetylase activity in either lytic or nonlytic cell-based formats and with extracts or recombinant enzymes. SAHA inhibition of K562 cells is shown.
Ultimately, advancing more HDAC inhibitors into viable therapeutics will require a more in-depth understanding, not only of the basic science of HDACs, but also their pharmacology, suggests Sriram Balasubramanian, Ph.D., senior director, translational research at Pharmacyclics.
“We are finding that HDAC inhibitors work well for hematological malignancies such as lymphomas, leukemias, and multiple myeloma, but only partially for solid tumors, which represent the majority of cancers. So, a basic question is ‘how do we get them to work here?’ It is likely that HDAC inhibitors may function best in combination with other therapies. But, to answer these questions, we will need to elucidate basic mechanisms of how HDACs function. As that pool of knowledge grows, we will be better able to determine how to combine HDAC inhibitor therapy with other strategies.”
Dr. Balasubramanian says another challenge is dealing with toxicity issues. “It is estimated that anywhere from 4–20 percent of the transcriptome can be altered as a result of using a single pan-HDAC inhibitor such as SAHA. Additionally, tens to hundreds of proteins may become acetylated after treatment with a pan-inhibitor. On the plus side, it appears that normal cells are more resistant to the apoptotic effects of HDAC inhibition. This may partly explain how clinical efficacy can be achieved at the tolerated doses.”
Since all of the HDAC inhibitors currently marketed or in clinical development inhibit multiple isoforms, broader applications of HDAC inhibitors will require a more sophisticated understanding of how each isoform works.
“There is clearly a role of specific isoforms in certain types of cancer, as well as in other diseases. But because of their similarity, it can be difficult to make selective compounds. This has been a real struggle in the field. Even with high-throughput screening, the progress has been limited by the structural information available,” Dr. Balasubramanian explains.
“Since the binding pockets of different isoforms can be quite similar, high-resolution crystal structures are needed. HDAC isoforms have been hard to crystallize individually, partly because they are usually found in complexes and with few exceptions, not as single entities. What is needed is more structure-guided rational design and computational chemistry.”
The exciting prospect of HDAC inhibitor therapeutics is clearly on the radar. Resolving specificity and toxicity issues should help launch even more candidates into clinical trials.
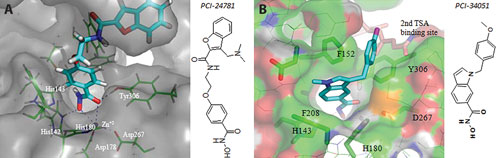
Binding of pan-HDAC and HDAC8-isoform-specific inhibitors: The pan-HDAC inhibitor PCI-24781 is shown bound in the long narrow pocket of a HDAC1 model (A), in contrast to the HDAC8-specific inhibitor PCI-34051 bound in the shallower pocket of HDAC8 (B). [Pharmacyclics]

