
March 15, 2006 (Vol. 26, No. 6)
Antibody Microarray Development on Ultrathin Film Nitrocellulose
Simultaneous analytical measurement of multiple protein biomarkers continues to hold promise in the identification of molecular signatures that may impact research applications, ranging from translational medicine to prognostic advancements, as well as early decision-making tools in drug discovery. Unfortunately, development of quantitative multiplex immunoassays is burdened with the task of selecting and characterizing binding molecules that deliver exquisite sensitivity, specificity, and precise measurements.
GenTel BioSciences(www.gentelbio.com) designs, develops, and validates multiplex immunoassays that provide up to 30 quantitative measurements per biological sample. The popularity of planar antibody microarrays is primarily driven by cost efficiency, minimal specimen consumption, and the opportunity to visualize the image quality of the processed immunoassay data.
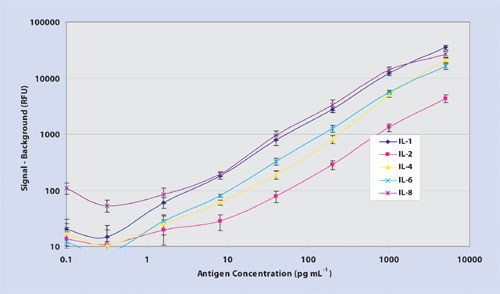
Figure 1a. 5-plex dose-dependent titration curves
Multiplex Immunoassay Sensitivity and Dynamic Range
Multiplex immunoassay development begins with a solid support. Surface chemistry lays the initial foundation in assay design and heavily influences the analytical results pertaining to sensitivity; defined here as the lowest concentration of measured analyte that can be detected with a 5% probability of erroneously detecting background signal in a defined system. Currently, there is no single comprehensive study that explains why microspot immunoassays consistently deliver better sensitivity than ELISA when compared using identical reagent sets.
Specific mechanisms that focus on analyte-ligand reaction kinetics likely involve surface chemistry effects such as the configuration of antibody immobilized on a matrix, nonspecific adsorption of proteins, as well as the optimal number of ligand-binding sites available after blocking the antibody printed support.
Antibodies bind to nitrocellulose non-covalently, minimizing the modification of antibodies previously well-characterized in ELISA applications that routinely fail when chemically coupled to latex microspheres as practiced with bead-based multiplex analysis. The mechanism of irreversible antibody attachment to thin-film nitrocellulose is likely due to the initial adhesion of immunoglobulin to the membrane surface through electrostatic interaction, followed by long-lasting attachment achieved by both hydrophobic interaction and hydrogen bonding.
Thin-film nitrocellulose provides a hydrophobic microenvironment, which may alter the hypervariable region of the antibody-binding site, thus providing a higher degree of access of the complementary determining region to bind an epitope. Importantly, microarray data processed on ultrathin film nitrocellulose using Cy3 fluorophore delivers minimal nonspecific background resulting in log-fold higher signal-to-noise ratios, improved sensitivity, and 45 logs of dynamic range (Figure 1a, Table). Moreover, thin-film nitrocellulose offers the low, reproducible analytical variance (Figure 1b) at both ends of the titration curve as expected in an ELISA test (<10% correlation of variation.)
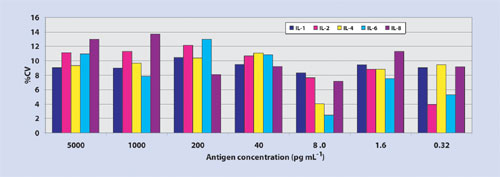
Figure 1b. Dose-dependent correlation of variation
Multiplex Immunoassay Specificity
Specificity is a critical test characteristic to prove multiplex immunoassay credibility. Confident assurance of immunoassay specificity hinges on repeated measurements of both minimal detectable dose and titration curve slope values for each analyte in the menu. Examples of cross-talk can be seen in either of the unwarranted results: nonspecific binding between the capture antibodies and an inappropriate antigen; nonspecific binding between the detector antibodies with an inappropriate antigen-capture antibody complex (Figure 2a); nonspecific binding between the detector antibody with an inappropriate capture antibody (Figure 2b).
Specificity testing between the antigen and the capture antibody is done by probing individual antigens against an array of capture antibodies followed by individual detector antibodies (Figure 2c). Specificity testing between the antigen and the detector antibody is analyzed by probing cocktails of antigens against arrays of capture antibodies followed by individual detector antibodies (Figure 2d).
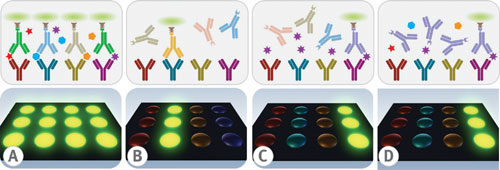
Fig. 2(a) nonspecific binding with an inappropriate antigen-capture antibody complex. (b) Nonspecific binding with an inappropriate capture antibody.(c)Probing microarrays with individual antigens and detector antibodies
Challenges of Protein Content and Validation
There are times when third-party commercially available antibody duo sets and/or purified antigens required for assay development are unavailable. In many circumstances, there may be multiple clones of commercial antibody available without access to antigen. Development of mouse monoclonal antibodies using purified native or newly developed recombinant antigen has become more affordable over the past ten years.
When working with newly developed immunoassay reagents, one must demonstrate and characterize that antibodies are capable of binding to purified as well as endogenous antigen within a complex heterogeneous mixture. Testing specific antibody binding to endogenous antigens is initially qualified through Western Blot analysis. Unless one is fortunate enough to access biological specimens from antigen-deficient cells, then you should remove endogenous antigen using immunoaffinity chromatography to separate antigen out of complex protein samples such as serum or plasma.
If successful, then the multiplex immunoassay of antigen-depleted samples should result in the absence of signal contrary to positive signal measurements observed when testing normal pooled real-world specimens. Validation for multiplex immunoassay performance ought to compare single-plex titration curves against multiplex titration curves to identify any interferences with levels of detection, precision variance, dynamic range, or slope.
There are many circumstances when matrix effects occur when testing plasma samples for some analytes, which leads to changes in the sensitivity and precision characteristics of the assay. A fivefold dilution of these samples in diluent buffer usually eliminates these interferences but sometimes may result in a lower absolute sensitivity linked to the dilution of plasma samples.
Not all antibodies perform well in multiplexed immunoassays. For example, there are antibody clones for TGF-b that require antigen denaturation using strong acid treatment. Additionally, some antibody clones derived from phospho-peptide immunization react only with the denatured phospho-specific protein target.
There are many examples where these immunoassays require a chaotropic agent to unfold and expose buried antigenic epitopes. Chemical treatments such as these typically obstruct immunoassay performance of other antigens and antibodies in the panel when using antibodies specific for the native antigen configuration.
Broad Applications
Multiplexed protein measurements have a wide range of scientific purposes related to virtually any disease where more than one protein is involved, including in processing a wide variety of body fluids, biopsied tissues, and other complex biological specimens. To name a few, we have measured coagulation proteins in citrated plasma samples, metabolic and inflammatory disease secreted proteins in serum, as well as cell-signaling pathway proteins in whole cell lysates.
Other applications of multiplex immunoassays have been in the search of a comprehensive profile of biomarkers that may identify surrogate endpoints in monitoring the benefits of therapy and in the recognition of adverse inflammatory profiles in the course of clinical trials, involving drug safety and efficacy. In summary, the benefits of multiplexing proteins are driven by more measurements with the goal of achieving higher quality data with less sample in less time.

