
Spatial genomics holds great promise in the quest to unravel the function of the multiple cell types in biological microenvironments. An array of technologies is being applied to understand these complex ecosystems in indications such as cancer, progressive neurodegenerative diseases, and cardiotoxicity. The overarching goal is to develop novel therapeutic approaches, whether they involve previously identified or newly discovered targets.
But analytical hurdles exist. Spatial technologies generate massive amounts of complex data, and user-friendly analysis tools are needed before the approach can become widely adopted. Experts came together at the Spatial Biology US, an online event organized by Oxford Global, to discuss how spatial biology platforms are incorporating multi-omics, imaging, and computational technologies to inform translational research and speed the development of new therapies.
Homing in on oncology targets
In the nascent field of spatial genomics, enabling technologies not only capture gene expression information but also retain the spatial context of tissue samples. These technologies range in their depth and resolution. Some technologies offer gene panels to support specific hypothesis testing; other technologies offer transcriptome profiles to support the de novo discovery of new disease targets and clinical correlates.
“Spatial genomics allows us to profile clinical samples reflecting the disease state in patients, including banked FFPE cohorts,” says Anna Lyubetskaya, PhD, Principal Scientist, Bristol Myers Squibb. To date, particularly in academic research, the few established applications focus on creating a spatial atlas with molecular tissue taxonomy and cell type localization targets.
“Since the technologies are resource intensive, require long specialized training, and leverage extensive cross-functional expertise, we need to be deliberate with our efforts,” cautions Eugene Drokhlyansky, PhD, Principal Scientist at Bristol Myers Squibb. “Interpretation of the outputs is nontrivial.”
The difficulties cited by Drokhlyansky are evident in the evaluation of tumors, which are complex ecosystems comprised of interacting cancer and noncancer cell types. “Even when we have access to patient samples, not all genomics technologies have the resolution to profile these disease systems,” Drokhlyansky remarks. “Profiling of disease is often obfuscated by the variety of cell types and spatial compartments in and surrounding the tumor tissue,” Lyubetskaya adds.
Characterization of tumor cells using single-cell genomics loses tissue context, and tumor purity is a confounder in bulk genomics data. Spatial genomics enables high-fidelity identification of the disease regions of the tissue to accurately profile the disease state and heterogeneity.
A particular strength is the ability to bridge the gap between genomics and pathology. “Pathology characterization of clinical tissue is the workhorse of patient care and late-stage drug development, whereas genomics is not yet as widely applied clinically,” Lyubetskaya elaborates. “But now we can characterize the same tissue using methods from both fields, which enables validation and integrative analysis between these key orthogonal readouts.”
“At scale, we can model cell-cell interactions and verify that cells are co-located in the disease, which was not previously possible,” Drokhlyansky asserts. This allows prioritization of the paracrine interactions that likely underlie disease for subsequent validation.
Analyzing microenvironments with modular tools
Spatial transcriptomics and proteomics technologies promise to provide detailed molecular characterizations of tissue architectures and cellular microenvironments. However, these technologies generate multiparametric data that is difficult to process and analyze with today’s computational platforms, says Darya Orlova, PhD, Senior Scientist, Genentech.
Currently, the analysis of spatially resolved omics data typically requires coding abilities. To remove the need for coding abilities, Genentech scientists are developing Spatial Expression Explorer (SPEX), a web-based image analysis application that accepts plug-in analysis modules. The application can execute various algorithms, include ones for cell segmentation, cell phenotyping, and the recognition of spatial expression patterns and cell-cell co-occurrence.
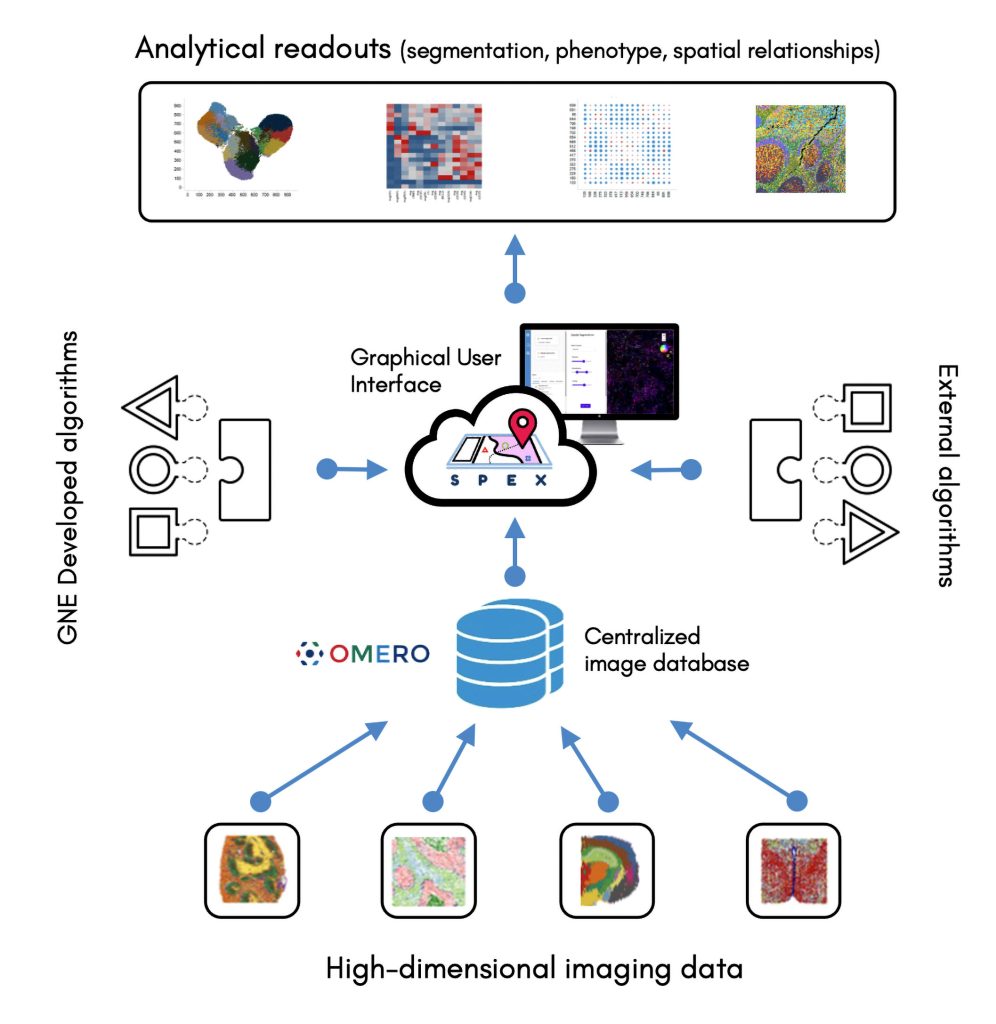
[Image courtesy of Genentech]
SPEX provides both infrastructure and quantitative analysis methods for streamlined access to open source image data management systems, such as the Open Microscopy Environment Remote Objects (OMERO) platform, along with preprocessing, analysis, and interactive visualization of spatial expression data. The application integrates multiple data modalities and leverages spatial analytics to couple single-cell RNA sequencing with other forms of analysis, such as evaluations of cell-cell communications between spatially distinct cell microenvironments.
In complex multicellular organisms, cells are organized into tissues that are in turn organized into organs, and so on. At each level of organization, the structure is closely related to the function that is governed by different gene expression programs.
“This suggests that if we could measure gene and protein expression of individual cells in a given tissue and be able to efficiently incorporate spatial cell arrangement information into these data, we would be able to infer, or at least hypothesize, function,” Orlova points out.
Recently developed technologies such as imaging mass cytometry, multiplexed ion beam imaging, co-detection by indexing (PhenoCycler™), and multiplexed error-robust fluorescence in situ hybridization (MERFISH) allow the acquisition of a spatial landscape of the proteome and transcriptome in individual cells. Orlova emphasizes that widespread adoption depends on the development of analytical capabilities and infrastructure to extract meaning from complex data.
SPEX was used to identify new tumor-immune microenvironment patterns in heterogeneous samples from a cohort of triple-negative breast cancer patients. In a study of idiopathic pulmonary fibrosis, SPEX was used to characterize the spatial arrangement of macrophage subtypes that emerged in response to disease. And SPEX was used to reveal mesenchymal signatures indicative of epithelial-mesenchymal transition. These signatures, which were found in cell communities on the periphery of a tumor, emerged in a xenograft model of human non-small cell lung cancer.
Characterizing the microglial response to amyloid plaques
Microglial cells are involved in neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. The cells are known to continuously survey, sense, and respond to their microenvironment within the brain and the central nervous system. They are also known to be highly sensitive to environmental changes and to demonstrate significant differences in aging and disease-related expression signatures between rodents and humans. Yet microglial cells are challenging to study in isolation. As a result, studies of microglial cells that could shed light on neurodegenerative diseases often resort to model systems.
“Model systems that recreate aspects of the disease microenvironment are important to understand and characterize the interaction of microglial cells with amyloid and tau pathologies that are observed in Alzheimer’s disease,” says Rebecca S. Mathew, PhD, Associate Principal Scientist, Merck Research Laboratories. “A chimeric mouse model with these pathologies provides important insights to how human microglial cells sense, respond, and interact with the tissue microenvironment.”
Spatial transcriptomics allows comparison of single-cell signatures to expression signatures that are derived from intact tissue sections. “We have identified differences in gene expression between human and mouse microglia within subregions of the brain, and we have used spatial transcriptomics to define expression signatures for cells that are proximal to regions showing characteristic Alzheimer’s disease pathology,” Mathew continues. “The technology also helps us to understand how human genetic targets alter microglial signatures and pathological states.”
Implementation challenges included embedding and sectioning tissues, ensuring consistent collection of sections (which must come from the same brain subregion across multiple animals within a cohort), handling and analyzing data, resolving both human and mouse expression signatures from the chimeric model datasets, and deconvolution of cell types located in proximity to Alzheimer’s disease pathology.
“We generally overcame challenges by establishing quality control tests for our tissues, standardizing protocols for isolating and banking tissues, and developing analysis pipelines to handle spatial transcriptomics datasets from chimeric model studies,” Mathew reports. “We are also working on strategies to deconvolute cell types that are located close to Alzheimer’s disease pathology.”
As the resolution of experiments increases, it will become possible to measure changes in gene expression with single-cell resolution and to observe cell-to-cell communication events using intact tissue sections. Mathew expects spatial transcriptomics technologies to be combined with cutting-edge multi-omics and histology techniques, such as RNA expression and protein sequencing, or REAP-seq.
Assessing the cardiotoxicity of oncological agents
Many cancer therapies have been clinically associated with detrimental cardiac effects. Unfortunately, these effects are poorly understood at the molecular level. For example, researchers have yet to clarify the molecular mechanisms behind antineoplastic drug–induced cardiotoxicity. Consequently, efforts to protect against this side effect have focused on risk detection and characterization rather than refinement of chemical design.
Molecular understanding offers an opportunity to develop improved mitigation approaches in drug development. Although it has been linked to both pathological and drug-induced heart failure, the role of intercellular signaling in cardiotoxicity remains to be understood (Guo et al. Trends Pharmacol. Sci. 2021; 42(8): 675–687).
Varied in silico, in vitro, and in vivo model systems are used during drug discovery and development to identify and mitigate potential cardiac safety issues. Throughout the drug discovery process, the applied models increase in complexity, enabling deep insights into potential cardiotoxicity risk prior to human trials.
Models range from cell lines overexpressing certain proteins, induced pluripotent stem cell–derived cardiomyocytes, advanced in vitro models (such as human cardiac microtissues composed of the three main cell types in the heart—cardiomyocytes, cardiac endothelial cells, and fibroblasts), to small and large animals.
“Use of in vitro models such as human cardiac microtissues enables us to mimic key aspects of cardiac biology, detect potential risks, and generate novel mechanistic insights,” says Amy Pointon, PhD, Senior Director, AstraZeneca. “By applying these innovative models and spatial technology, we are discovering new biological insights that allow us to understand why some therapies cause adverse cardiac effects. This knowledge can be used to develop improved approaches.”
AstraZeneca is focused on producing robust methods to generate spatial understanding in in vitro models, such as microtissues, which have a low biomass and require optimization and refinement. These methods are now being applied to derive new insights into the heart and the cell signaling processes within.



