Epigenetic modification of RNA, the RNA analog of the DNA epigenome, has been observed for over 40 years and is known as the epitranscriptome. Messenger RNAs (mRNAs) in particular are post-transcriptionally modified, obtaining base modifications as well as 2ʹ methylation of sugars near the 5ʹ cap. Cap modifications identify the mRNA as self rather than foreign, and modulate stability, translation, decapping, and regulation of mRNAs. Tools to effectively mimic the epitranscriptome have not existed until now. With the development of a co-transcriptional capping method called CleanCap®, mRNAs with each of the four different types of 5ʹ caps can be produced. In this article, we describe this new technology and its implications for mRNA therapeutics.
Messenger RNA is a popular alternative platform for gene replacement, genome editing, immuno-oncology, and vaccines, compared to plasmid and DNA viral vector–based therapeutics. DNA-based vectors can integrate randomly into genomes, activating undesired genes such as proto-oncogenes. These insertional mutagenesis concerns notably surfaced when children with “bubble boy” disease developed leukemia when their gene therapy lentiviral cassette inserted adjacent to, and activated, a proto-oncogene. This spurred an interest in alternative technologies such as mRNA.
Messenger RNAs cannot integrate. Therefore, concerns of insertional mutagenesis are diminished. Messenger RNA expression is usually more transient than that of plasmids or viral vectors. Its applications include genome editing (such as CRISPR/Cas9, zinc finger nucleases, and TALENs), gene replacement, immunotherapeutics (such as CAR T-cells and bispecific antibodies), and RNA vaccines. However, therapeutic development requires understanding the interactions of mRNA with cellular machinery. Much like DNA, mRNA contains epigenetic marks that affect its function. An ideal synthetic therapeutic mRNA should mimic fully processed mRNA and not activate innate immune pathways. For mRNA vaccines, however, innate immune stimulation may be desired.
An introduction to mRNA capping
The mRNA 5ʹ cap regulates splicing, nuclear export, resistance to exonuclease degradation, and translational initiation. The cap also marks the mRNA to the innate immune system as “self RNA.” The eukaryotic 5ʹ cap consists of a 7-methylguanosine (m7G) connected by a triphosphate bridge to the first nucleotide, forming a structure known as cap 0. Cap 0 is sufficient to recruit translational initiation factors and prevent mRNA degradation. Methylation of the 2ʹ-hydroxyl group of the first cap-proximal nucleotide then creates cap 1. In ~50% of transcripts, an additional 2ʹ-O-methylation of the second nucleotide forms cap 2. Frequently, the first nucleotide is adenosine, which can be N6 methylated to form m6Am cap. Thus, in addition to the cap 0 intermediate, there are at least three endogenous cap structures found in mature eukaryotic mRNAs: cap 1, cap 2, and m6Am cap (Figure 1).
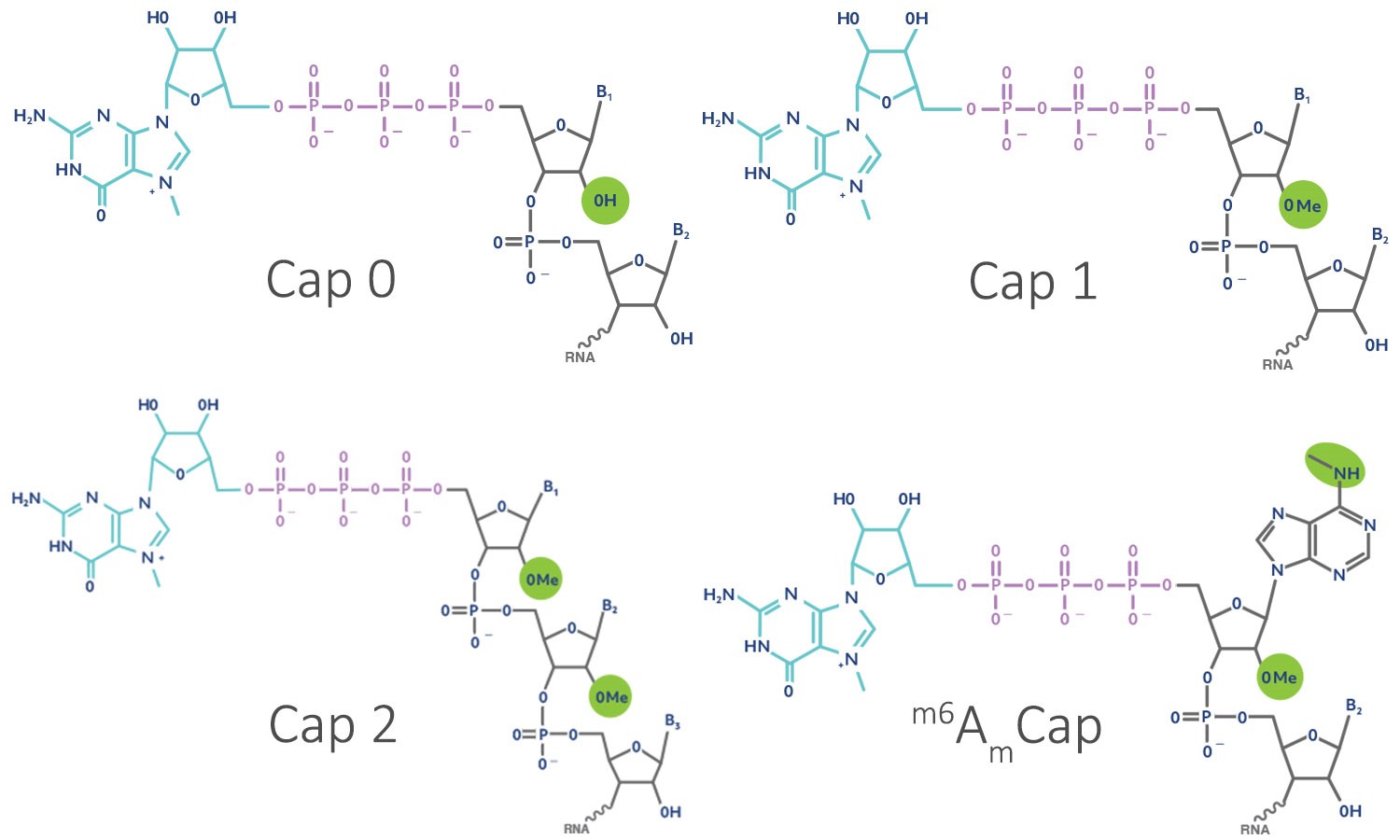
Cap 1 and cap 2 were discovered in the 1980s, but their function remained largely unknown. Cap 1 structures are frequently found in cytoplasmic viruses. Revealingly, deletion of the viral methyltransferase responsible for cap 0 to cap 1 conversion results in attenuated viruses, suggesting an important role for this epigenetic mark. Several studies have linked this attenuation to recognition by the innate immune system, which screens for molecular patterns unique to pathogens. Host immune proteins such as RIG-I and IFIT recognize aberrantly capped RNAs. RIG-I recognizes uncapped 5ʹ-triphosphate RNAs or cap 0. Intriguingly, cap 1 modification abrogates RIG-I signaling. IFIT proteins recognize improperly capped RNAs lacking 2ʹ-O-methylation and bind their 5ʹ ends. IFITs prevent translation of viral sequences by competing with translational initiation factors. Thus, cap 1 structures may authenticate mRNAs as “self RNAs.” Accordingly, cap 0 RNAs show very little activity in vivo relative to cap 1 RNAs (Figure 2). The function of cap 2 remains unclear, despite the fact that about half of all mRNAs possess a cap 2 structure.

Recently, m6Am cap has gained increased attention. Roughly 30–40% of mRNAs possess this new cap form that is thought to engage specialized translational initiation factors to enhanced translation. The m6Am capped RNAs are also more resistant to microRNA silencing and are more stable in cells. This may be due to decreased decapping by the enzyme Dcp2.
Manufacturing capped mRNAs
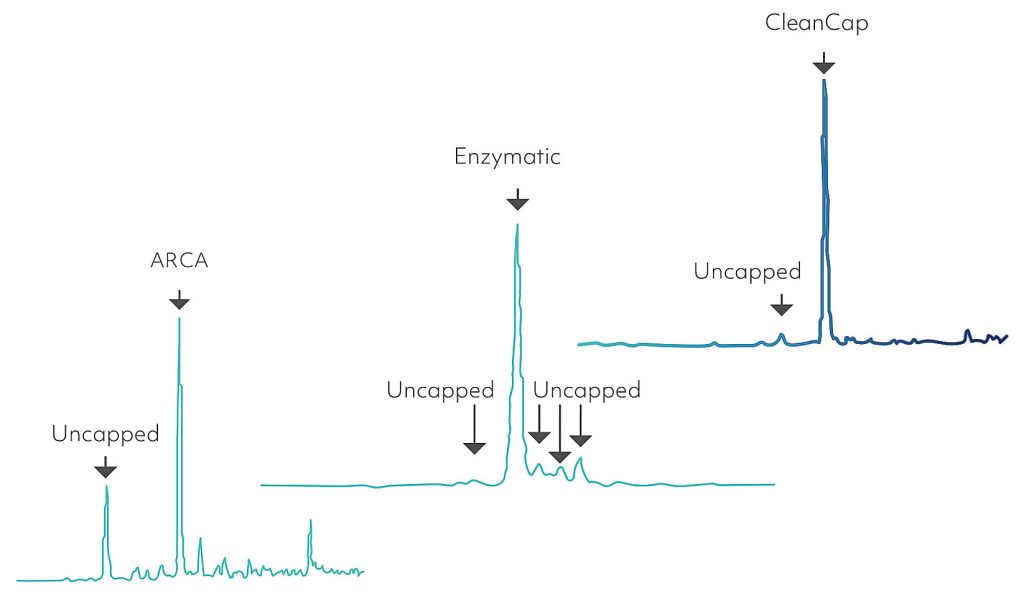
There are two main ways to produce capped RNAs in vitro. The first is to cap the RNA after the transcription using vaccinia virus capping enzymes, which results in cap 0 or cap 1 RNAs. Capping can be highly efficient, but it is variable and expensive at scale. Enzymatic capping cannot produce cap 2 or m6Am cap. Alternatively, RNA can be capped during transcription using an excess of a cap analog. Until recently, the most popular cap analog was known as ARCA (anti-reverse cap analog). ARCA results in immunogenic cap 0; however, capping is inefficient (~70% capped/30% triphosphate). Yields with this legacy capping method are low (~1.5 mg/mL of transcription).
A new co-transcriptional capping method called CleanCap (TriLink BioTechnologies) has seen rapid adoption recently. In contrast to ARCA, which uses a dimer (m7GpppG) to initiate T7 transcription, CleanCap initiates using a trimer (m7GpppAmG). While T7 RNA polymerase has a strong preference for GTP initiation resulting in enzymatically or ARCA-capped RNAs beginning with a 5ʹ-guanosine, CleanCap in combination with T7 polymerase is flexible with the identity of the 5ʹ nucleotides, therefore allowing initiation with a variety of 5ʹ sequences. This method produces highly capped RNAs with improved yields (~4 mg/mL of transcription). Importantly, the CleanCap method can yield cap 0, cap 1, cap 2, or m6Am capped mRNAs. Figure 3 compares capping with these three different methods.
The future of mRNA therapeutics
Self-replicating RNAs are an exciting new vaccine development platform generally derived from alphaviruses. The viral structural genes are replaced by an antigenic payload, allowing the RNA to replicate without forming infectious virus. Replication of the RNA increases persistence and activates innate immune sensors that recognize the double-stranded replication intermediate, leading to robust immunization. One challenge in the production of alphavirus-based vaccines is that the 5ʹ end of the alphavirus begins with 5-m7GpppAU. Using previous methods, producing this type of cap would not be feasible. But with CleanCap, which allows for the production of alphavirus vectors with an authentic 5ʹ end, it is.
Messenger RNA therapeutics are now entering the clinic, and generating safe mRNAs that mimic fully processed, properly capped, and authenticated mRNA will be essential. Many initial trials focus on ex vivo treatment of cells before returning them to the body to minimize safety concerns. These programs utilize mRNA-dependent genome editing to insert chimeric antigen receptors or correct an underlying defect such as sickle-cell anemia or b-thalassemia. There are also numerous mRNA-based vaccine trials including self-replicating RNAs designed to vaccinate against infectious agents or tumors. More ambitious trials seek to deliver mRNA to humans directly to treat diseases such as muscular dystrophy or urea disorders. Generation of properly capped mRNAs will be critical for the success of this new therapeutic platform.




