
Over 80% of U.S. drug prescriptions are for generic medicines.1 Such medicines are an important part of the medical system, helping to alleviate drug shortages and improve access to affordable treatment options for patients, clinicians, and healthcare providers.
Generic drugs must provide the same clinical benefits as reference listed drugs (RLDs) and fulfill strict FDA standards. They must demonstrate “bioequivalence” to an RLD in pharmaceutical, biological, and therapeutic terms. Their active ingredients, dosage forms, routes of administration, strengths, and related properties must be the same (pharmaceutical); their active pharmaceutical ingredients (APIs) must be similarly available and active at the site of action (biological); and the drug must offer a similar safety and efficacy profile in a clinical setting (therapeutic).
For simple, orally administrated drugs, bioequivalence is evaluated via competitive blood level trials, as the therapeutic agent must enter systemic circulation to reach the site of action. However, it is far more challenging to demonstrate bioequivalence for complex generic drug products (CGDPs), which represent complicated formulations or routes of administration and, therefore, must typically be verified using expensive and lengthy clinical endpoint studies. This challenge has grown in the disruption caused by the COVID-19 pandemic, with many clinical trials halting and with many drug developers seeking alternative ways to progress their product portfolios.
However, methods for verifying CGDP bioequivalence are also available in the form of in vitro bioassays, which can be used in augmented testing workflows to prove bioequivalence to regulators. An integrated bioassay-based approach can support biowaiver applications, enabling in vitro data to be used in lieu of clinical data. Whereas flexibility is always valuable in drug discovery, the versatility afforded by biowaivers is crucial for pharmaceutical companies looking to weather stochastic events and bring complex generics to market amid COVID-19-driven trial shutdowns.
An integrated approach to bioequivalence
The biomorphological, physical, and structural characteristics of a drug’s dosage form directly influence the in vivo parameters of a formulation, including the rate and extent to which the drug is delivered to the target site. CGDPs possess complex APIs, formulations, delivery routes, drug-device combinations, and dosage forms, vastly increasing the difficulty of bioequivalence analysis.
Additionally, a generic may have a similar formulation to an RLD but perform differently in vivo, especially given the amount of intersubject variability within a reference population. Whereas physicochemical characterization (also known as Q3 testing) can establish comparability between CGDP formulations, these in vitro tests cannot definitively predict in vivo outcomes.
To meet regulatory requirements, drug developers require a totality of evidence on a formulation’s characteristics and behavior in vivo to confirm therapeutic equivalence to an RLD. For this, integrated approaches—in which bioassays are used alongside existing tests—have proven effective at characterizing not just a single formulation property, but the overall behavior of a multifaceted formulation in a biological setting.
Such an approach enables drug developers to select the most biorelevant system or assay with which to assess CGDP performance, and to evaluate the combined impact of discrete physicochemical characteristics in a multifaceted way. Augmenting existing workflows with in vitro bioassays can reveal whether changes in the microstructure of a complex formulation will impact critical performance parameters in vivo, explicitly connecting API and formulation to biological effect to improve confidence in and understanding of a given formulation.
The effects of formulation: Ocular and topical products
Accurate, sensitive, reproducible bioassays—sometimes augmented by biorelevant models—can evaluate postulated mechanisms of action driven by an API and/or other formulation-specific characteristics. Two examples are presented here to illustrate the process that may be used to demonstrate CGDP equivalence.
The impact of formulation can be demonstrated by TobraDex® 1%, an antibiotic ophthalmic suspension that treats inflammation in the eye. Increased retention time is known to improve bioavailability, so this strategy is often used in ophthalmic products.2,3
TobraDex® was prone to settling, limiting its efficacy, so a new formulation was developed. The new formulation, called TobraDex ST® has half the API strength and uses xanthan gum as an excipient to decrease sedimentation and increase residence time on the ocular surface. After application, the formulation mixes with tears; this alters the product’s ionic content and pH and drives a tobramycin-gum interaction that raises viscosity, enabling the product to reduce inflammation to the same degree as TobraDex® 1%.
Using the “in vitro permeation test” (IVPT), studies conducted by Absorption Systems showed that the new formulation had higher corneal flux and greater corneal tissue association, whereas an in vivo pharmacokinetic rabbit model showed equivalence between the ST and 1% formulations of TobraDex. This, in turn, matched associated clinical bioequivalence data, demonstrating that bioassays such as IVPT could be used as a surrogate for clinical bioequivalence studies.
Similarly, IVPT assays have proven sensitive in evaluating how formulation differences affect percutaneous absorption of topical products. An IVPT study evaluated the absorption of drugs based on a novel Versatile Cream Base and compared these to the RLDs.4
Each formulation was applied to human cadaver trunk skin mounted onto Franz diffusion cells. Over the following 48 hours, serial samples were then taken of any permeated drug in the receptor chamber. Compared to the RLDs, the Versatile Cream Base products delivered significantly greater (2.3- to 2.4-fold) amounts of permeated drug, demonstrating enhanced percutaneous absorption and highlighting the potential for improved transdermal delivery systems for analgesic medicines via improved vehicles.
A surrogate for in vivo
The FDA is increasingly including in vitro options in its product-specific guidance for CGDPs, indicating regulatory openness to such approaches in the development of complex drugs.5,6 To verify CGDP bioequivalence, developers must incorporate biorelevant tools and assays into their testing, with Q3 testing used to characterize formulation function and bioassays to characterize a product’s mechanism of action as part of a complementary “augmented Q3” workflow. Such an augmented approach is especially important in the currently disrupted pharmaceutical landscape, in which drug developers must seek alternative ways to verify bioequivalence (Figure 1).
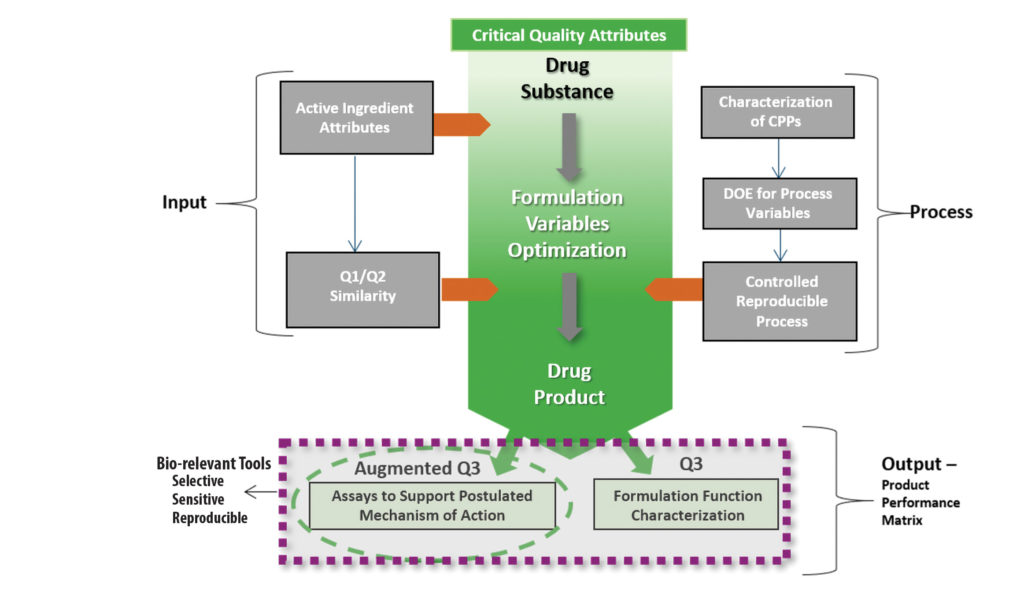
In vitro experiments hold a number of benefits over in vivo trials: they shorten development timelines, lower costs, and raise fewer associated ethical or safety considerations. They are also appropriate for products that may be highly variable, may have severe side effects or addiction and abuse potential, or must be administered to patients rather than healthy volunteers.
Biowaivers for orally dosed drugs are evaluated based on the FDA’s Biopharmaceutics Classification System (Figure 2)7; this requires knowledge of product permeability, intestinal solubility (both related to APIs), and dissolution (related to formulation).
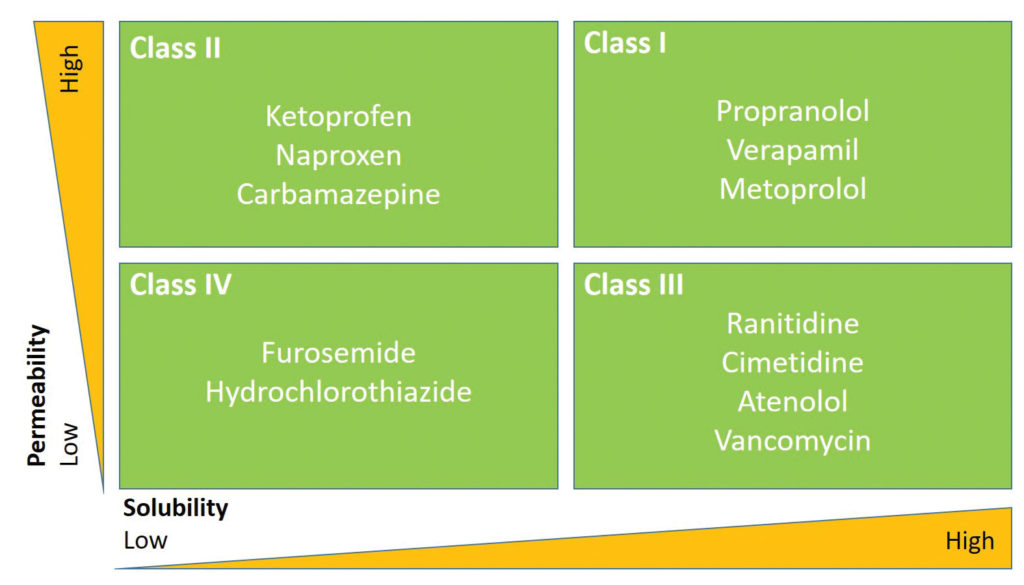
For locally acting CGDPs, biowaivers must be supported by orthogonal in vitro measurements—which incorporate early, intermediate, and extended formulation-controlled aspects of performance—and can be supplemented by in silico modeling. The collective weight of evidence from multiple such approaches replicates the regulatory process of RLD approval and supports the evaluation of equivalence in human efficacy, thereby giving confidence to regulators, clinicians, and patients.
Bringing CGDPs to market quickly, safely, reliably
The pharmaceutical industry is seeking accurate, efficient, and reproducible ways to assess CGDP bioequivalence. Integrated and augmented testing using advanced bioassays can avoid the roadblocks to CGDP development.
Using multiple testing approaches with complementary strengths mitigates the risks of individual testing methods, and can streamline regulatory approval, especially given that in vitro options for CGDP bioequivalence testing are increasingly supported by the FDA. In vitro bioassays can evaluate the combined impact of discrete physicochemical characteristics and assess whether formulation differences are biologically meaningful in vivo, supporting biowaiver applications and accelerating the progress of CGDPs to market.
References
1. Nash DB. The Use of Medicines in the United States: A Detailed Review. Am. Health Drug Benefits 2012; 5(7): 423.
2. Patel A, Cholkar K, Agrahari V, Mitra AK. Ocular drug delivery systems: An overview. World J. Pharmacol. 2013; 2(2): 47–64.
3. Scoper SV, Kabat AG, Owen GR, et al. Ocular distribution, bactericidal activity and settling characteristics of TobraDex ST ophthalmic suspension compared with TobraDex ophthalmic suspension. Adv. Ther. 2008; 25(2): 77–88.
4. Wang X, Black L. Ex vivo percutaneous absorption of ketamine, bupivacaine, diclofenac, gabapentin, orphenadrine, and pentoxifylline: comparison of versatile cream vs. reference cream. Int. J. Pharm. Compd. 201317(6): 520–525.
5. Food and Drug Administration. FDA Statement: Statement from FDA Commissioner Scott Gottlieb, M.D., on 2019 efforts to advance the development of complex generics to improve patient access to medicines. Released: January 30, 2019. Accessed: August 2, 2020.
6. Davit, BM, Kanfer, I, Chung Tsang, Y, Cardot, J-M. BCS Biowaivers: Similarities and Differences Among EMA, FDA, and WHO Requirements. AAPS J. 2016; 18(3): 612–618.
7. Benet LZ. The role of BCS (biopharmaceutics classification system) and BDDCS (biopharmaceutics drug disposition classification system) in drug development. J. Pharm. Sci. 2013; 102(1): 34–42.
Vatsala Naageshwaran, ([email protected]) is chief business officer at Absorption Systems.




