
February 1, 2015 (Vol. 35, No. 3)
Mark Hughes Ph.D. Senior Product Manager Thomson Reuters
Understanding Disease Mechanisms through Multi-Omics Data Integration Pathway Analysis
Advancements in next-generation sequencing (NGS) technologies have enabled researchers to generate genome-wide data of unprecedented quality and quantity. Genomics, expression, microRNA, chromatin IP, methylation, histone modification, and more recently chromosome confirmation capture are rapidly moving into the clinical setting. Projects like 1000 Genomes, Encode, Blueprint, and many smaller projects are providing a rich source of background information and understanding of the genome and the epigenome in relation to normal and disease states.
These applications and content are utilized in numerous research fields from early disease understanding and patient stratification through preclinical and clinical applications like precision medicine. With data generation growing at an exponential rate, the need for efficient analyses, data reduction, and comprehensible visualizations is critical for biomedical interpretation of NGS data.
In the precision medicine age companies are trying to identify specific variants within a group of patients that are then targeted in the drug development pipeline. Identifying these patient cohorts has been greatly advanced by NGS analysis; however the scale, volume, and diversity of data possible with cohort studies and multiple omics approaches all present new challenges. From just handling these large files to identifying the significant data out of the hundreds of millions of data points generated, a systems approach is needed to combine it and aid in identifying driver mutations of a disease.
Thomson Reuters has enhanced the scope of data and types of analysis needed with MetaCoreTM, an integrated software suite for functional analysis of NGS, variant, CNV, expression, metabolic, SAGE, proteomics, siRNA, microRNA, and screening data to include a variant analysis workflow within the system.
Looking at the NGS pipeline, from raw files to biomedical knowledge, starts with the alignment of raw reads and subsequent analysis of the dataset to compare them to reference or a control/s. Following this are application-specific steps, e.g., variant analysis or expression analysis.
The first steps of the pipeline, demanding in terms of processing power and storage requirements, can be automated and pipelined for high-throughput analyses. Turning this output into meaningful results, however, requires interpretive downstream analysis steps to layer the biological meaning around the data. To achieve this, a significant amount of high-quality background information at several different levels is required.
For example, to annotate regions of relevance, a comprehensive genome annotation database must be available to allow correlations with multiple types of genomic elements such as genes, proteins, metabolites, compounds, drugs, transcripts, transcription start sites and/or regulatory regions (promoters/enhancers) and their interactions. MetaBaseTM contains such annotations as well as information about direction, effect, trigger, tissue, and other contextual information.
After mapping data to a comprehensive database combining these annotations, merging results like RNA expression, proteomics, metabolomic, and genomic variant analysis can give detailed insight into molecular processes, e.g., proteins encoded by a specific transcript variant or even structural variants of the genome causing diseases like cancer.
The genomic analysis tools inside MetaCore enable the filtering of variant data with 21 different filters including functional predictions, conservation scores, population frequencies, protein domains, and all MetaCore proprietary ontologies, while also extending results to the full functionality of MetaCore pathways, enrichment analysis, network building, and interactome analysis to elute mechanisms of the disease or differences in patient groups.
These data types rarely act on their own; integration into a network or pathway system is a reasonable subsequent move to understand the disease mechanism. Variants can be combined with gene/transcript expression, CNV, proteomic, or metabolic data and used in all MetaCore workflows to find connections in these data types (Figure 1).
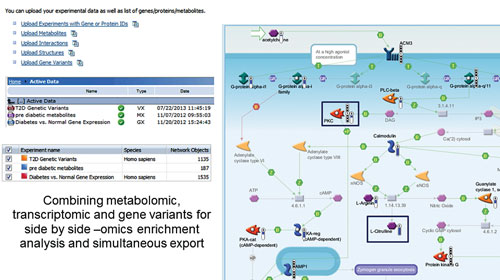
Figure 1. Workflow results combining variant, gene expression, and metabolite datasets
Case Study
This case study showcases MetaCore functionalities through filtering a colorectal cancer (CRC).vcf file, running a pathway map enrichment with the prioritized variants as well as incorporating CRC transcriptomic data from the MetaCore microarray repository. Multi-omics approaches have already proven to be powerful tools in uncovering disease phenotypes with limited sample size, including n = 12. Here, we propose multiple hypotheses around the mechanisms of CRC using a combination of knowledge and data driven approaches available within MetaCore.
MATERIALS AND METHODS
The genomic data1 was obtained comparing a CRC and a normal sample and initially filtered using the following settings in MetaCore: 1) Variant Classification: Type; Single, Functional Class; Missense or Nonsense, 2) Functional Prediction: Logistic Regression Prediction; Damaging or Absent, LRT; Deleterious or Absent 3) Novelty: 1000 genomes; Present or Global (gMAF) (Freq: 0.001) or Absent 4) Disease: Colorectal Neoplasms.
Transcriptomic data was obtained from the MetaCore microarray repository (Human Colorectal Cancer vs. Normal Adjacent Colon comparison) following extraction from ArrayExpress (E-MTAB-572), quality control, and filtering the DEGs fold changes > ± 1.5 and p value < .05.
RESULTS
Variant filtering resulted in 10,167 variants mapping to 1,054 network objects that were used for enrichment. The most significant map folder was colorectal neoplasms (p value = 1.068e-39) indicating the genes that play an important role in CRC pathological signaling. To understand the relationship with colorectal cancer signaling at the mRNA level, the filtered E-MTAB-57 transcriptomic data was compared side-by-side within the map folder ontology. One map, “Inhibition of calcitriol/VDR signaling in colorectal cancer,” is shown in Figure 2. Through map analysis and map overview content, the following hypotheses are proposed to explain CRC specific mechanisms from the genomic and transcriptomic data:
- CYP27A1 (three variants post filtering) catalyzes conversion of colecalciferol into calcifediol,3 which is converted in to calcitriol by another enzyme.4 Calcitriol signaling is downregulated in CRC; therefore these CYP27A1 mutations could represent loss of function mutations that decrease catalysis of colecalciferol and overall decreased VDR signaling.
- The CRC genomic data shows a variant in ZO2 (TJP2 gene) at position 71865988 with a C>T SNP and the transcriptomic data shows the TJP2 gene to be upregulated 1.8 fold (p value = .021). This variant could be hypothesized to lead to increased production of TJP2 and change biological processes such as CRC progression through cell adhesion mechanisms.
- This map describes the cell cycle driving cell proliferation, which feeds into the CRC phenotype. The data shows two variants in cyclin-dependent kinase inhibitor 1A or p21 (positions 36651971 C > A and 36652122 C > G), which could be involved in dysregulation of the cell cycle leading to changes in cell proliferation. This map also shows upregulation of c-Myc at the mRNA level (fold change = +2.3 and p value = .0009), which could also contribute to increased cell proliferation. Therefore this combination of genomic and transcriptomic changes could be hypothesized to drive CRC through dysregulated cell proliferation.
These three hypotheses represent a fraction of those needed to further investigate mechanisms of colorectal cancer progression. Through combining filtered genomic and transcriptomic data, pathway analysis allowed identification of potential metabolomic signaling consequences and biological processes potentially dysregulated in CRC. In conclusion, analyzing multi-omics data in a pathway analysis context provides a powerful method for generating systems biology hypotheses.
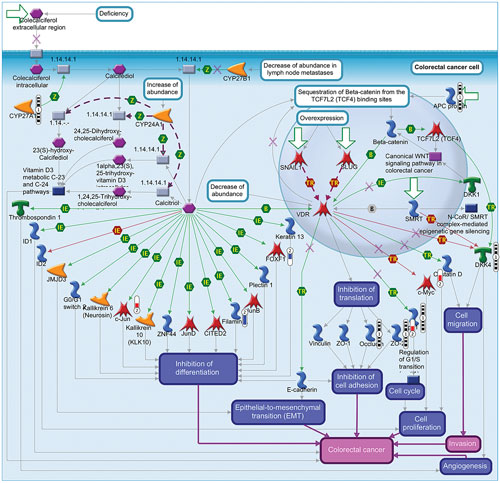
Figure 2. Pathway map demonstrating inhibition of calcitriol/VDR signaling in colorectal cancer (source: MetaCore)
Mark Hughes, Ph.D. ([email protected]), is the senior product manager, systems biology at Thomson Reuters.
References
1 PMID:24312117
2 PMID:16919171
3 PMID:9210654
4 PMID: 19667168





