
August 1, 2015 (Vol. 35, No. 14)
Andrew Anfora Ph.D. Senior Field Application Scientist BioNano Genomics
Saki Chan Research Associate BioNano Genomics
Alex Hastie Ph.D. Research Team Leader BioNano Genomics
Michael G. Saghbini Ph.D. Director of Assays and Reagants BioNano Genomics
Boudewijn F.H. Ten Hallers Associate Scientist BioNano Genomics
Purification of Chromosomal-Length Plant DNA Molecules
Eukaryotic genomes are organized into a finite number of linear double-stranded DNA molecules (chromosomes) ranging in length from kilobases to gigabases and containing unique and nonunique segments. To assemble a genome, DNA is extracted from cells and prepared, and data is generated in the form of overlapping fragments (reads) and assembled into a set of contiguous sequence fragments (contigs). When two unique regions are separated by a nonunique region that is longer than the relative sequence read, the unique regions cannot be linked, and the result is a fragmented contig or incorrect assembly.
Next-generation sequencing (NGS) techniques produce sequence reads that are typically no longer than 200 bases, which is often too short to resolve three common types of large-scale nonunique materials: tandem repeat arrays, segmental duplications, and homologous regions of heterozygous chromosomes.
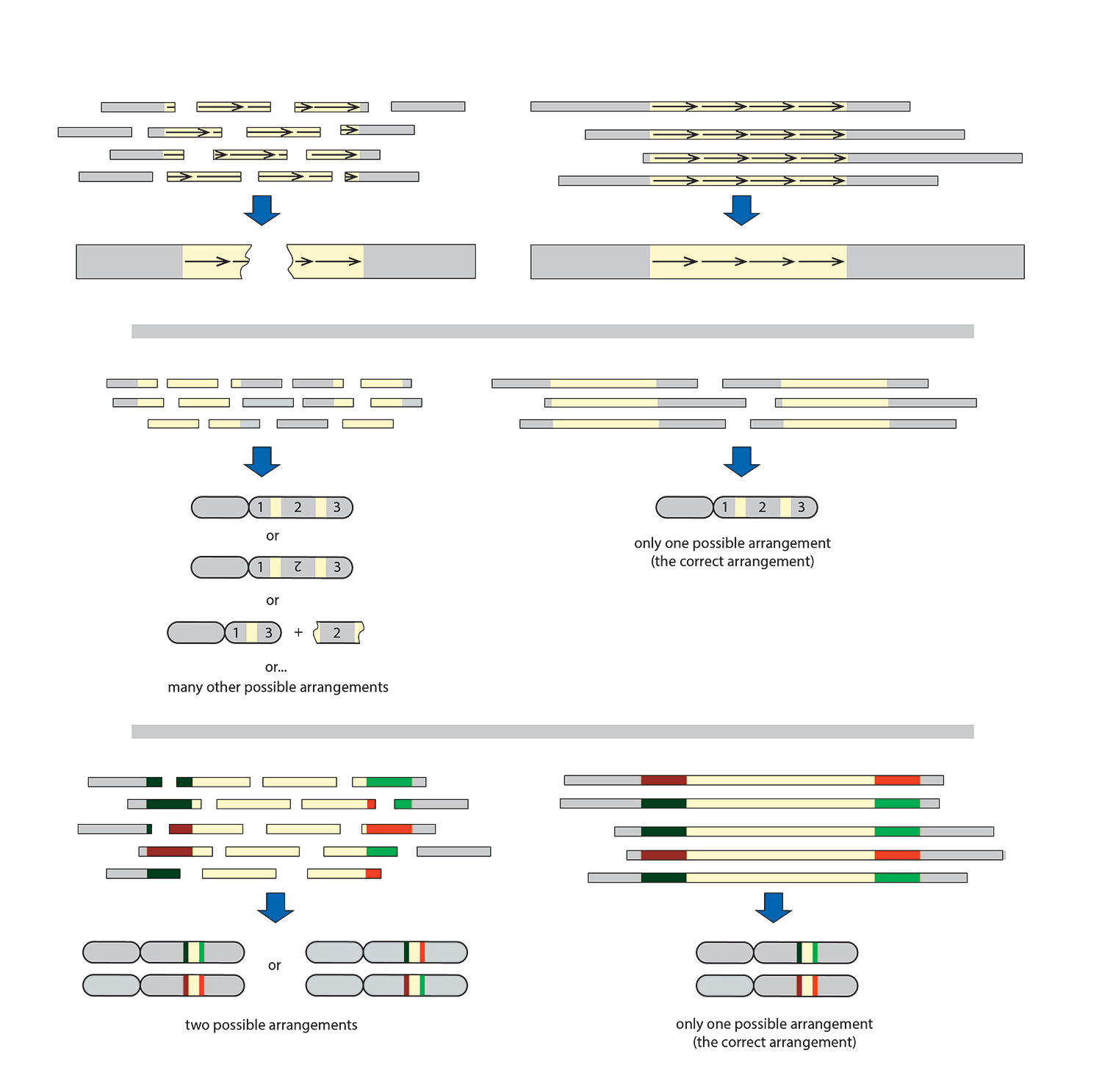
Tandem repeat arrays are stretches of DNA with repetitive sequence ranging from a few base pairs to multiple megabase pairs. A sequence read cannot span the entire array, and it is therefore impossible to get the information needed to determine the number of times the repeat motif occurs (copy number) (Figure 1A).
Segmental duplications are long segments (1,000 to 400,000 base pairs) of DNA that occur more than once in a genome (on the same or different chromosomes). A sequence read that does not span the entire duplicated portion does not provide the information needed to connect the two unique flanking sequences that are used to help identify if the sequences are from different or the same chromosome (Figure 1B).
Heterozygous chromosomes are two or more nearly identical chromosomes that have variants that may specify different traits on each copy. A sequence read that does not span the entire shared region between the variants does not provide the information needed to determine if the heterozygous chromosomal variants are on the same or different DNA strands (phasing), resulting in loss of variant information (Figure 1C).
Figure 1. Comparison of short and long reads and their resulting assemblies. For an unambiguous assembly, reads must contain unique elements flanking both ends of nonunique segments. Unique sequence: gray; nonunique sequence: beige. (A) Tandem repeat arrays consist of repeated elements (arrows). They are abundant in most genomes, ranging from a few to thousands of copies in one or multiple arrays. (B) Segmental duplications are low copy, interspersed repetitive elements that are highly homologous. (C) Heterozygous chromosomes are nearly identical chromosomes with variants on different strands. The process of defining whether variants are in cis (on the same strand) or trans (on different strands) arrangement is called phasing.
Confront Problematic Regions
Newer mapping and sequencing technologies have longer read lengths and are quickly becoming powerful tools for resolving the problematic regions since longer read lengths are able to span the tandem repeat arrays, segmental duplications, and the regions between variants of heterozygous chromosomes (Figure 1, rightmost panels; Figure 2), placing the bulk of the challenge instead on the quality of the starting material.
To utilize the newer mapping and sequencing technology, DNA isolation techniques must advance from beyond the current isolation preps that enrich for DNA fragments 100 to 30,000 base pairs in length, while continuing to address the challenges that accompany the complex cell architecture of plants. Following the recommendations provided here can help to achieve extraction of plant chromosomal DNA over 500,000 base pairs long.
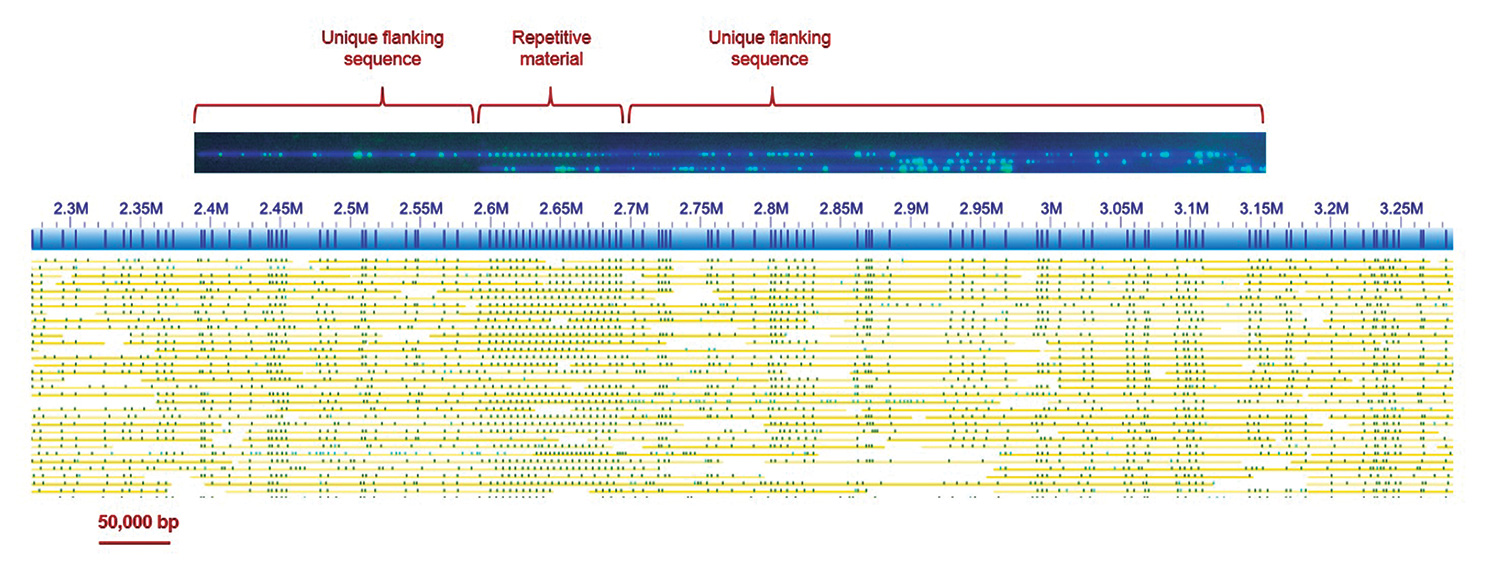
Figure 2. Top: An example of a single molecule of plant DNA imaged after being elongated in a NanoChannel Array on the BioNano Genomics Irys® System. DNA molecules were labeled at a sequence motif with a fluorescent dye (green) and then stained with an intercalating dye, YOYO-1 (blue). The DNA was then linearized in NanoChannels and imaged. Bottom: A 1 Mbp segment of a genome map de novo assembled from such single molecule data. Consensus map: blue; labels: dark blue vertical stripes; single DNA molecules used to construct the consensus map: yellow lines with green spots. Of particular interest in this example is the region with 19 evenly spaced labels, with 4.7 kbp interval units. The even spacing of motif sites correlates with tandem repeat arrays, in this case spanning 89.3 kbp. To correctly assemble such a repeat array and associate adjacent unique genomic regions, single reads must span the array and anchor uniquely on each side.
Enrich for Clean Nuclei
The single most important factor for successful isolation of intact chromosomal-length plant DNA is to use biomass with the highest DNA content-to-mass ratio possible and the least amount of metabolites, generally very young, dark-treated tissue. The ideal starting materials are seedlings that have been dark-treated for 24–72 hours. This will not be practical for all plants; however, the next best starting materials are leaves that have just finished budding and are still young tissue. Aged plant tissue should be used only as a last resort. If a bolus of plant tissue is not available in a single collection, snap-freezing the tissue and holding it at −20°C or below also works well.
The first unique biological obstacle to isolating plant DNA is the cell wall. The cell wall must be fractured without damaging nuclei to minimize genomic DNA fragmentation. The most efficient way to accomplish this is to first briefly fix the tissue in formaldehyde to stabilize nuclei and then mechanically disrupt the cell wall by mincing or briefly homogenizing with a rotor-stator homogenizer.1 The resulting crude nuclei suspension is then treated with a mild detergent, Triton X-100, to differentially lyse mitochondria and chloroplast; filtered to remove cellular and extra cellular debris; and enriched by centrifugation.
Throughout the nuclei isolation process, specific chemicals are used to preserve DNA integrity while removing unwanted contaminants. High pH, EDTA, and salt are used to minimize nuclease activity. 2-Mercaptoethanol (βME) and polyvinylpyrrolidone (PVP) are used to control polyphenols. PVP is a water-soluble polymer that binds polyphenols in order to prevent polyphenols from binding to the DNA; 2-mercaptoethanol prevents their oxidation.
If polyphenol oxidation occurs while the polyphenolic compounds are bound to DNA, an irreversible binding event takes place, producing DNA that will not respond to enzymatic treatment as expected. PVP-10 and PVP-40 can be used interchangeably; an 8% PVP solution is usually effective for removing polyphenols.
Use a very high concentration of βME (7.5% v/v) in both the isolation buffers and in the first step of the Proteinase K digestion of agarose-embedded nuclei. When working with βME, it is important to remember to work in a hood, wear appropriate personal protective equipment, and use fresh (<6 months old) βME.
Besides polyphenols, polysaccharides need to be controlled. Polysaccharide plant inhibitors can be removed from crude nuclei preps with extensive washes. Even with extensive washing, many plants, especially dicot species, will benefit from Percoll® (GE Healthcare) gradient or cushion cleanup. Percoll is a suspension of silica particles that have been coated with PVP. In addition to further binding polyphenols, a Percoll density gradient or cushion will also help to remove particles of a different density than the nuclei.
As an alternative to Percoll purification, fixed nuclei can be separated from debris by use of flow cytometric sorting, the gold standard for nuclei purification.
Obtain High-Quality DNA
After the nuclei are sufficiently cleaned, they are embedded in low-melting-point agarose plugs for removal of detergent-soluble material and proteins. After lysis and protease treatment, plugs should be washed and equilibrated in high EDTA buffer. Following solubilization of agarose with β-agarase, and after drop dialysis, it is possible to achieve pure, chromosome-length DNA.
This DNA can be used in many molecular mapping and sequencing methods. It should, however be visually inspected. Plugs should appear clear, although sometimes small amounts of polysaccharide contamination can produce slight opacity.
These recommendations provide principles and methods to extract some of the longest and highest quality plant DNA. Current and future long-range DNA analysis platforms should benefit from implementing these principles and methods whenever high performance is expected.
1 Šimková, H., Cíhalíková, J., Vrána, J., Lysák, M. & Doležel, J. 2003. Preparation of HMW DNA from plant nuclei and chromosomes isolated from root tips. Biologia Plantarum 46: 369–373.
2 Zhang, M., Zhang, Y., Scheuring, C., Wu, C., Dong, J. & Zhang, H. 2012. Preparation of megabase-sized DNA from a variety of organisms using the nuclei method for advanced genomics research.
Nat. Protoc. 7: 467–478.
3 Lam, E., Hastie, A., Lin, C., Ehrlich, D., Das, S., Austin, M. & Kwok, P. 2012. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 30: 771–776.
Andrew Anfora, Ph.D. ([email protected]), is a senior field application scientist, Saki Chan is a research associate, Alex Hastie, Ph.D., is research team leader, Michael G. Saghbini, Ph.D., is director of assays and reagents, and Boudewijn F.H. Ten Hallers is an associate scientist at BioNano Genomics.





