
Source: Cell Metabolism
Scientists in Australia say their research points to fat tissue as a source of disease, and widens our understanding beyond the traditional focus on liver and pancreas as the main culprits. The study (“Protein Kinase C Epsilon Deletion in Adipose Tissue, but Not in Liver, Improves Glucose Tolerance”) appears in Cell Metabolism.
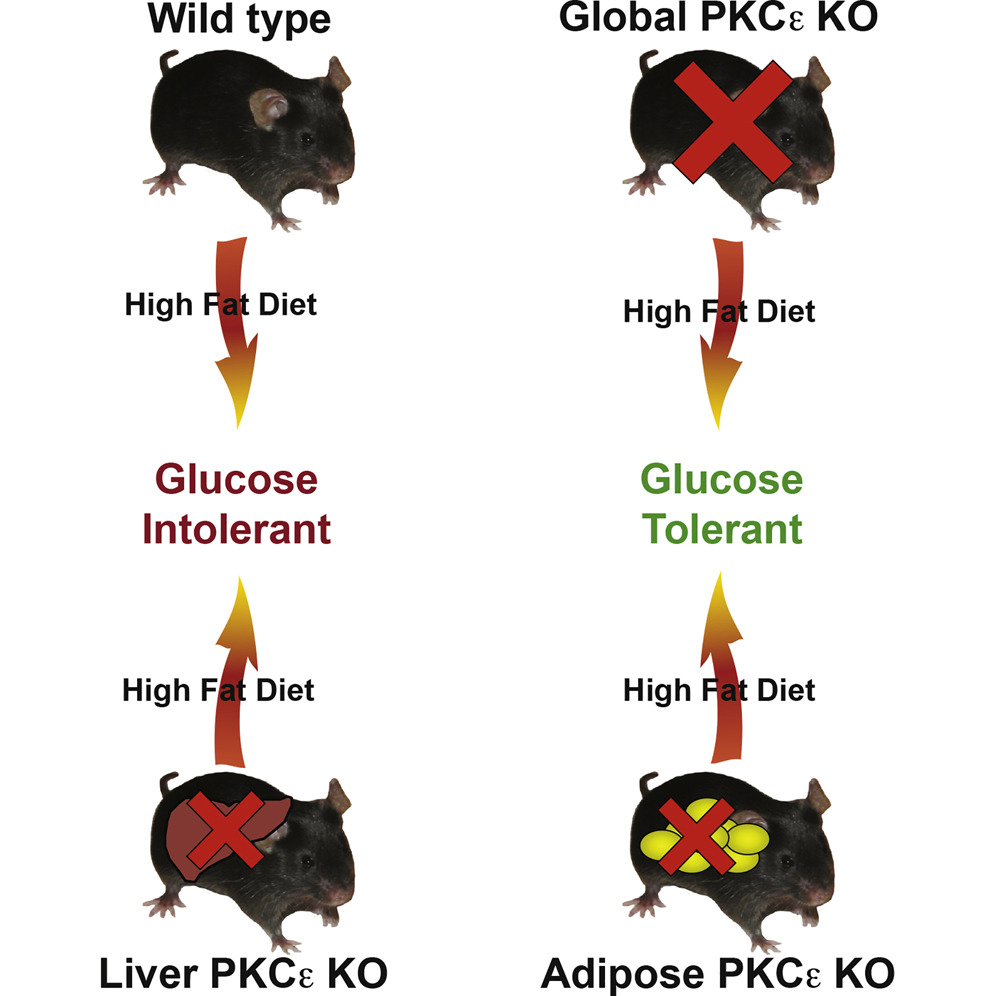
“Protein kinase C epsilon (PKCε) activation in the liver is proposed to inhibit insulin action through phosphorylation of the insulin receptor. Here, however, we demonstrated that global, but not liver-specific, deletion of PKCε in mice protected against diet-induced glucose intolerance and insulin resistance. Furthermore, PKCε-dependent alterations in insulin receptor phosphorylation were not detected. Adipose-tissue-specific knockout mice did exhibit improved glucose tolerance, but phosphoproteomics revealed no PKCε-dependent effect on the activation of insulin signaling pathways. Altered phosphorylation of adipocyte proteins associated with cell junctions and endosomes was associated with changes in hepatic expression of several genes linked to glucose homeostasis and lipid metabolism,” write the investigators.
“The primary effect of PKCε on glucose homeostasis is, therefore, not exerted directly in the liver as currently posited, and PKCε activation in this tissue should be interpreted with caution. However, PKCε activity in adipose tissue modulates glucose tolerance and is involved in crosstalk with the liver.”
The new research is centered around the surprising finding that PKCε, known to be involved in diabetes, isn't acting in the liver or the pancreas as was once assumed. Researchers have long known that PKCε is important for the development of diabetes. Mice that have no PKCε produced anywhere in the body don't develop diabetes-like symptoms, even under conditions where other mice become diabetic.
“We have known for some time that if you remove PKCε entirely from mice and feed them a high-fat diet they do not become glucose intolerant. In fact, they are protected from becoming diabetes-like,” says associate professor Carsten Schmitz-Peiffer, Ph.D., Garvan Institute of Medical Research.
Diabetes researchers use a high-fat diet (HFD) to induce type 2 diabetes in mice. Following a HFD, most mice become glucose intolerant—they are unable to control their blood sugar after eating. In particular, the liver becomes insulin resistant and no longer responds to the pancreatic hormone insulin.
“The big surprise was that when we removed PKCε production specifically in the liver, the mice were not protected. For over a decade, it's been assumed that PKCε is acting directly in the liver. By that logic, these mice should have been protected against diabetes,” notes Dr. Schmitz-Peiffer. “We were so surprised by this, that we thought we had developed our mice incorrectly. We confirmed the removal and tested it in several different ways, but they still became glucose intolerant when given a HFD.”
If not the liver, then where? The team's goal was to determine where PKCε was working to progress glucose intolerance.
“What we found,” explains Dr. Schmitz-Peiffer, “is that if we removed PKCε production solely from fat tissue, the mice were protected from becoming glucose intolerant, similar to when we removed PKCε from the entire animal. So PKCε isn't progressing diabetes from the liver, but in fact, it is acting from fat tissue to worsen the disease.”
PKCε has been known to be expressed in multiple tissues, but Dr. Schmitz-Peiffer's findings now point to a new function in fat tissue. A close examination of the fat tissue revealed a striking difference in the shape and size of the fat cells in the presence and absence of PKCε.
“Under the microscope, the fat cells looked very different,” Dr. Schmitz-Peiffer says. “In HFD-fed mice with PKCε removed from the fat tissue, we saw mostly small, healthy fat cells. And in HFD-fed mice with PKCε intact, which are glucose intolerant, we saw more of the unhealthy, engorged fat cells, that tend to have less access to oxygen and become inflamed.”
For Dr. Schmitz-Peiffer, it is clear this could have wide-ranging and complex implications for diabetes.
“We know that fat tissue is a lot more than just an inert mass for storing fat,” he explains, “it's a very dynamic organ, it sends many messages and releases factors that communicate with the rest of the body, including the liver.
“If PKCε is changing the nature of fat and affecting the overall health of fat cells, it's changing the types of messages it sends and factors it releases, which could be acting on the liver and possibly other organs to interfere with glucose metabolism.”
Ten years ago, Dr. Schmitz-Peiffer and his team showed that removing PKCε entirely protected mice against glucose intolerance. “We just never knew where PKCε was acting. But we knew we might be onto something therapeutically important if we could find a way to block PKCε”.
Today, he is collaborating with colleagues at the Fragment Based Drug Discovery Platform at the Monash Institute of Pharmaceutical Sciences to develop an orally available peptide that can disrupt PKCε activity.
“These results give us an even better idea about how to target PKCε to develop the most effective treatments possible. And therapeutically targeting PKCε would be a new possible approach for diabetes treatment.”





