
John Sterling Editor in Chief Genetic Engineering & Biotechnology News
Moving Forward with an Early-on Strategy
SPONSORED CONTENT BROUGHT TO YOU BY CORNING
Cell therapy has become one of the fastest-growing segments in the life sciences. It involves the delivery of whole, living cells to a patient for the treatment of disease. Typically, this category is divided into two main sections: autologous (meaning the cells are derived from the patient, modified [usually genetically], and re-administered back to the patient), or allogenic (meaning the cells are derived from another individual and show some match to the potential new host—similar to a blood transfusion).
GEN recently interviewed leading experts in the field to find out what considerations are critical in order to carry out a successful and efficient cell therapy manufacturing program. GEN’s cell therapy panel included:
- Derek Adams, Ph.D., chief technology and manufacturing officer, bluebird bio
- Robert Langer, Sc.D., is the David H. Koch Institute Professor in the department of chemical engineering at the Massachusetts Institute of Technology
- Richard Eglen, Ph.D., vice president and general manager, Corning Life Sciences
- Bruce L. Levine, Ph.D., is the Barbara and Edward Netter Professor in Cancer Gene Therapy at the University of Pennsylvania Perelman School of Medicine
- Shashi Murthy, Ph.D., professor of chemical engineering and director of the Sherman Center at the Northeastern University’s College of Engineering
- David Sourdive, Ph.D., executive vice president, technical operations, Cellectis
GEN: What do you see as a major pain point for cell therapy manufacturers during the scale-up process?
Dr. Adams: In autologous cell therapies, the major pain point is that one cannot really scale up any unit operation. The process is one patient, one batch—which limits batch volume and the associated economies of scale at that level. The small volumes and larger number of batches force replication of operations such as fluid movements, mixing, sampling, and labeling for every batch.
Dr. Langer: For autologous cell therapies, the major pain points are: 1) patient-to-patient variability in the starting material, 2) having multiple manufacturing spaces on line to increase throughput, and 3) variability in the overall yield of the final cell product. These factors, in addition to the complexity of current therapies, result in long process times and high costs that can limit their scalability and broad applicability across indications. For allogeneic cell therapies, the pain points are more similar to those of traditional biologics and scaling up the batch sizes of the various unit operations.

Derek Adams, Ph.D., bluebird bio (left) and Robert Langer, Sc.D., Massachusetts Institute of Technology (right)
Dr. Eglen: There are different challenges depending on the nature of the cell therapy, and the production process needed (see sidebar below “Cell Therapies Scale More Efficiently with Dissolvable Microcarrier-Based Cell Culture”). Cell-based therapy approaches fall into two categories: autologous or allogeneic. Most immunotherapy approaches, based on CAR-T technology, are autologous and consequently require low bioproduction volumes. In fact, most CAR T-cell cultures will be conducted in volumes less than 10 L. The challenge, therefore, lies in the ability to have maximum flexibility in the cell culture platform to accommodate the heterogeneity in the type and number of cells available initially from the patient. For example, after isolation and selection of the cells, some patients may have 100 million T cells while other patients have more than one billion cells.
Dr. Levine: The pain point has been having the appropriate reagents, materials, and equipment that can do the manufacturing at the scale of billions of cells and/or in a clinically compatible, FDA-compliant fashion. In the beginning, meaning a few decades ago, there were not the materials, reagents, and equipment that there are now. Apart from the materials and equipment is the fact that when you are doing research experiments, you are using cell lines. And if you are not using cell lines, you may be using primary cells; but those are generally from healthy donors. When you begin to work with cells from donors with disease, whether, for example, it is cancer or HIV, those cells look different in terms of their phenotype and composition and they behave differently. They are generally more challenging.
Dr. Murthy: The number one pain point is translating the process either from the academic bench to a large-scale environment or from a trial environment to commercial manufacturing. The recent news reports from Novartis regarding some challenges in the manufacturing process for KymriahTM reflect this challenge. Originally approved for pediatric acute lymphoblastic leukemia (ALL), the reports describe how Novartis has encountered challenges for the more recently approved second indication, diffuse large B-cell lymphoma, where the manufactured product was found to be ‘out of spec’ because the commercial label specification was more stringent than in prior clinical studies.
Academic medical centers have become [more] sophisticated in recent years, and they use a lot of advanced equipment, while partnering with tool companies, as well. So scaleup is not necessarily a question of resources or access to advanced tools. The primary challenge is the reliance on manual processes during the research phase, where there is inevitably (and necessarily) a lot of tweaking various steps on the fly, and the transition to a commercial manufacturing where entire processes need to occur within very tight parameter limits. This transition is the number one challenge.
Dr. Sourdive: From the perspective of Cellectis, our experience is largely focused on genetically engineering T cells, especially for adoptive T-cell immunotherapy for cancer. We are developing and manufacturing allogeneic, off-the-shelf CAR-T cells as our main product line.
The product is a cell population. It is a cellular mix. Even if the cells are quite alike, there are nuances and differences. We think that is one major pain point: unavoidable heterogeneity in cell populations, including in starting cell material.
Scaling up is not just ‘more of the same.’ It is about asking something slightly different of the cells. So there are some qualitative things to be taken care of while the scale-up process is taking place. That is one important pain point.
Another pain point pertains to cost and resources. With cell therapies, especially those based on human primary cells, there is always a limitation in the amount of material. Some players, especially for autologous approaches, decide to scale out. But the cost is also quite important because manufacturing cells in batches for every individual can make it very challenging to get the costs down. This is less challenging with allogeneic approaches.
We would like to reemphasize that we are talking about a biological product which is difficult to control. For scaleup in the CAR-T field, we would also point out the extensive monitoring and control needed due to the biological nature of the product. When you are dealing with cell therapy, a major pain point is monitoring all of the parameters that affect cellular growth. That is really a big stumbling block in the field.
However, we would not want to give too bleak a picture because, even though these are pain points, we think the technological approaches that are now being deployed are getting better at handling these, especially with allogeneic approaches, because there is a lot more control that can be exercised on the starting material and the final product itself.
Healthy donor material is usually much better. It can be selected and can be required to abide by certain criteria, so there is probably less dispersion and less variability compared to autologous, patient-derived material.

Corning® Dissolvable Microcarriers were designed to provide a solution for simplifying the downstream processing and separation steps affiliated with microcarrier culture.
GEN: Are there currently any regulatory hurdles that slow the cell therapy scale-up progression, and how are manufacturers dealing with them?
Dr. Adams: The biggest regulatory hurdle relates to the overall understanding of our processes by the entire industry. Because we are still in the early days of developing and commercializing these therapies, we have a tremendous amount to learn about process behavior and the real relationships between product quality attributes and clinical safety and efficacy. As such, we all over-sample and over-test our products to collect as much data as possible to get at those questions.
For example, in gene and cell therapies that use lentiviral vectors for gene addition, we test every batch of vector and the transduced cell product for replication competent lentivirus (RCL). The vector manufacturing process is carefully designed to ensure production of infectious, but replication incompetent, virus. However, we as an industry do not yet have a large enough dataset and process history of producing negative RCL results to convince ourselves and regulators that the expensive and slow testing can be eliminated.
Dr. Langer: A big hurdle is the need to manufacture in Grade B clean rooms. Manufacturers are trying to make their processes fully enclosed to ensure the aseptic nature of their process in order to move their process into less stringent clean room spaces.

Richard Eglen, Ph.D., Corning Life Sciences (left) and Bruce L. Levine, Ph.D., University of Pennsylvania Perelman School of Medicine (right)
Dr. Eglen: More positive clinical trial data is now being reported. In addition, the number of regulatory approvals from the FDA of cell therapies is accelerating (See table on Approved Cellular and Gene Therapy Products). This rapidly changing environment is driving innovation in cell therapy research and development to address diseases, some of which have hitherto been untreated. In turn, this drives the demand for innovative technology solutions from companies, such as Corning, as well as the need to increase manufacturing capacity of existing products.
Dr. Levine: The question to ask is, ‘Are those regulatory hurdles appropriate?’ and ‘Do they guarantee or safeguard the development of new therapies that are as safe as they could be, given the risk-benefit ratio?’ The FDA is generally in a pretty good place and we have found the agency to be a collaborative partner. I know that may be different from some people's impression of the FDA as putting up roadblocks. Sometimes that could be the case. But with data and the appropriate scientific background, we have found the agency generally amenable within the scope of what the regulations and the guidelines are.
The FDA has been adapting to this field, especially under the current commissioner, Scott Gottlieb, M.D. Just a couple of weeks ago, the FDA issued six new guidances on cell and gene therapy products and that reflect an evolution of the field. If we think about hurdles, the obstacles are found in the Code of Federal Regulations and in the way they were written for small molecules and, to some degree, for conventional biologics where the raw material is the same every time.
When you have a cell therapy where the raw material is always different in an autologous product, that creates some challenges. When you have a cell product where the cells divide after administration, that is a dividing drug. How do you dose and characterize and develop a potency—an in vitro potency assay for a dividing drug where the potency is in vivo? Those are both scientific and technical challenges that we can work on together. But the regulations are what they are. They were written at a time when development of these cell products was far, far off beyond the horizon.
Dr. Murthy: I do not think that there are regulatory hurdles per se. I see the challenge being more with respect to seamlessly meeting quality and consistency requirements in manufacturing, which then relate to broader issues of process transfer and different levels of research (i.e., research to clinical trials, and then clinical trials to commercial manufacturing). The process, at that point, really includes the entire process from sample collection and then all of the unit operations’ downstream logistics (such as cryopreservation and thaw) until delivery to the patient. This is a very complex collection of steps.
I don’t believe that regulatory considerations are constraining cell-therapy scaleup. It is the complexity and large number of steps and associated quality and consistency requirements that are the challenge and the fact that we don’t yet have fool-proof toolkits that can guarantee the desired outcome for the entire process.
If you want to make an analogy to protein bioprocessing, we have standardized scale-up processes. You have your small bioreactors, larger bioreactors, and then you have scaleup. Process development and scaleup is a highly organized and well laid-out effort. But it has taken over 20 years to get there. By contrast, we are only in the early stages with cell therapy.
Dr. Sourdive: A regulatory hurdle for those who are in the manufacturing arena is having devices/reagents that can be labeled GMP grade, or having a drug master file available so that manufacturers can access these files and make their submissions, like INDs for example, easier for adoption. Cell therapy manufacturing may involve numerous materials and equipment, and suppliers know this is a hurdle for us manufacturers and are cooperative in getting these next steps available to us.
We also need to have some convergence of the way different agencies address issues and of what they put the focus on. We already see that there are nuances between different jurisdictions. Some things are more important in the U.K., while others are more critical in the U.S., and still others are more important in other European countries. That is normal. It is something we expect as these are still early days in the field.
But from a purely regulatory perspective, going toward more shared standards and agreeing and converging on the most important manufacturing, preclinical and clinical aspects will help. We think it is all going to be data-driven and the lessons learned from the experience we and others in the field are building both in manufacturing and in the clinic.
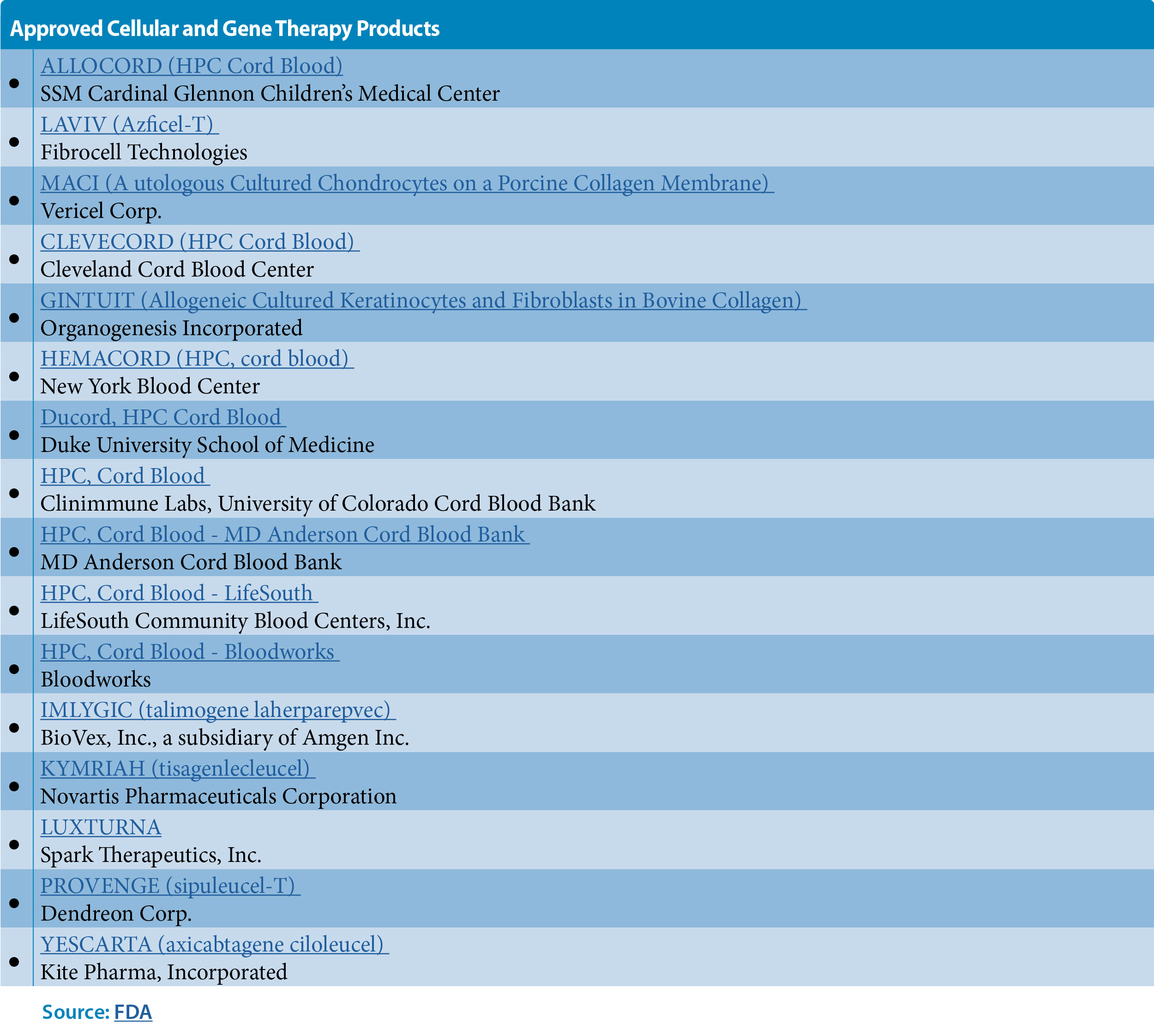
Table: Approved Cellular and Gene Therapy Products. Click here to access the pdf.
GEN: Have there been recent technological or scientific advancements that will have a significant impact on cell therapy manufacturing?
Dr. Adams: For autologous cell therapies, there are many closed system and automated technology solutions being developed that will provide incremental improvements to process robustness, reproducibility, and sterility assurance. These technologies may also reduce labor needs per batch helping to reduce costs. However, a truly safe, effective, and low-cost allogeneic therapeutic approach would be a game changer for the industry. There are many companies pursuing that approach, so we will see what the future brings.
Dr. Langer: One recent, exciting advance has been the SQZ delivery technology. This relatively simple, physical method allows one to deliver virtually any type of material to any cell type. This process has not only demonstrated the ability to engineer cell types/functions that are not accessible by other technologies but also maintains normal cell function very effectively compared to the more damaging existing technologies. Additionally, other technologies that allow the process to be shortened and to reduce the number of unit operations/process steps would be beneficial. For example, the ability to isolate the target cells of interest from blood or leukapheresis product in a single, rapid step would be impactful.
Dr. Eglen: A major challenge in cell therapy manufacturing today is the worldwide shortage of production capacity for viral vectors, critical to the manufacture and scale up of many cell-based gene therapies. Cell therapy manufacturers, including contract manufacturing organizations (CMOs), need to increase the yield of viral vectors in the most efficient manner possible, particularly in as small a footprint as possible.
Several years ago, Corning introduced our High Yield Performance (HYPER) technology, which utilizes a gas-permeable film as a culture surface, in a single-use, stackable vessel. This product allows robust gas exchange for the cells in culture in a small footprint. For example, a 36-layer HYPERStack® vessel is roughly the same size as the Corning 10-layer stacked vessels, but it provides more than three times the yield of adeno-associated virus (AAV) and lentivirus vectors.
Undoubtedly, other technologies will be developed that continue to address the need to culture cells in high abundance and viability. One area we are watching closely is the potential convergence of 3D cell culture technologies with bioproduction. It is now clear that cells grown in 3D systems mimic the physiological environment of cells in vivo. This has consequences for clinical research, drug development, and potentially for cell therapy manufacturing.
Dr. Levine: Gene-editing techniques have been a major step forward. CRISPR has been hot in the news but we do not want to overhype it. We did the first-ever gene-editing human trial not with CRISPR but with ZFN (zinc finger nucleases). So being able to directly knockout or insert, along with the ability to use viral vectors, and potentially without the need for viral vectors, those are huge advances. We are seeing advances in the design of receptors and combination receptors, as for example, with bispecific CAR-T cells and even trispecific CAR-Ts. So designing engineering strategies, including receptor designs, that enable cells to traffic, turn on, turn off, and be resistant to tumor-induced immune suppression is another huge step forward for the cell therapy field.
Dr. Murthy: There is a lot of activity with respect to cell therapy manufacturing and technology. The recent news from Torque Therapeutics with respect to their Slipstream™ platform is a great example. This is interesting for a couple of reasons. One is that this is a therapeutics company which made a significant investment in an in-house engineering team to create its manufacturing process. And it is not just a process: It is instrumentation. There is a lot of proprietary innovation and technology there. Also, in its announcement, the company makes it clear that its vision is toward decentralized manufacturing and that is a paradigm shift that is slowly coming into focus in cell therapy manufacturing.
The vision of cell therapies that can be manufactured with automated black-box-type instruments at a hospital is compelling. This greatly simplifies manufacturing and logistics. However, separately from that, we now live in an era of smart systems, e.g., systems capable of collecting massive amounts of data and then reacting to that in some form and then continuing to gather more data and reacting some more. That sort of mindset has now become an expectation with respect to the manufacturing of pharmaceutical products and not just cells. There are discussions like ‘Why can we not gather a large quantity of process data, and then for every batch of cells try to relate what happens in the patient to what happened in the process?’ (for example: response time or magnitude related to proportion of therapeutically active cells, related in turn to the quantity of starting material and how it was processed).
Indeed, there are some efforts going on in that regard that will yield a lot of innovation. But I should hasten to add that this is really hard stuff. Everything about this is difficult, from knowing what to sense to building the right kind of sensors. New types of sensors, for example, single-use and advanced data gathering and analytics capabilities are needed. Then there are broader questions of how the data is handled and what type of regulatory framework governs it.
The last point I want to make on this is that there is a lot of innovation involving the biology that is associated with cell therapy. For example, the development of allogeneic cell therapies and potential synergies with gene editing, new constructs and types of T cell and other therapies, potential reductions in dose sizes, and combinations of cell therapies with biologics, to name only a few areas. The manufacturing community will need to keep abreast of these developments and adapt our offerings accordingly, which will not be trivial.
Dr. Sourdive: One thing that is already transformative and will continue to be so is gene editing. Now, for the first time, we can control the genetic makeup of a cell or a product. We can engineer features in the product that will be beneficial both to the patient and to the manufacturing operations.
We already have examples of that as we brought gene-editing to the clinic with the CAR T-cell product line we are developing. There are some particular traits that are enabled through gene editing, by knocking out genes. The first breakthrough enabled by gene editing was making non-alloreactive T cells, allowing the development of off-the-shelf allogeneic products where T cell were traditionally used in autologous settings essentially as a graft. We also enabled T cells to be compatible with treatments (antibodies or chemotherapies) that are given to cancer patients: two of our UCART products under IND are gene-edited to resist alemtuzumab. Another example is allowing the use of tumor-specific targets that happen to be naturally present on the surface of T cells. If we want to make a CAR-T cell that is directed against such antigens, we can do so with gene editing, avoiding T-cell fratricide killing, and that is the case of one of our product candidates called UCARTCS1.
We have been a gene-editing company since inception. We saw it coming. We have been working on using gene editing to make things allogeneic, off-the-shelf, but also to add all these supraphysiological traits and features to the cells, allowing them to be better killers, to do what normal T cells cannot do and eventually circumvent the defense mechanisms of cancer. An example we disclosed was knocking out checkpoint inhibitors such as PD1 in T cells to prevent tumor-driven immune blockade. It is just the beginning. We think it is the key to deploying CAR T-cell therapies in solid tumors and will also apply beyond adoptive T-cell immunotherapy. Gene-edited designer cells or tissues will apply to regenerative medicine and other indications.
We would like to point out that next-generation sequencing (NGS) tools help make sure that gene editing is becoming more precise and targeted. We need to be able to identify all the effects that gene editing has on the genome of the cells.
The discovery of biomarkers has also helped us find out where gene editing can become a useful tool in demarcating areas where we can have some flexibility.
In addition, bioreactors have rapidly evolved in the area of cell therapy. They have allowed us to expand and proliferate these gene-edited cells.

Shashi Murthy, Ph.D., Northeastern University’s College of Engineering (left) and David Sourdive, Cellectis (right)
GEN: We often hear about scaling up vs. scaling out. Are companies currently leaning in one direction and do you think that is likely to change?
Dr. Adams: See my response to question one. For autologous cell therapies, the principle of scale out is really the only option.
Dr. Langer: The approach will really depend on the type of cell therapy. For autologous cell therapies, one has to scale out and implement multiple manufacturing lines at approximately the same scale. For allogeneic cell therapies, increasing the batch size and scaling up is the usual approach.
Dr. Eglen: The common constraint for both approaches is physical space—the process area, room footprint, and incubator space. Scaling out may be faster and simpler to implement than scaling up, but it requires much more space, which comes at a premium. On the other hand, scaling up better utilizes the available space and therefore scales more efficiently. Scale up limits the choice of vessel size but better accommodates the demand for customized configurations of the system. Demand for technologies that increase output and reduce footprint, such as HYPERStack, is increasing precisely because of its ability to provide both scale and customization. As the field moves to allogenic therapies, the demand for suspension culture scale up solutions will increase significantly due to the higher volume of cells required.
Dr. Levine: When I think of scaling up, I think of going from a 5 L to 5000 L bioreactor. In the case of autologous cell therapies, that is not needed since the dose requirements are set for one person, and we are scaling out with a current process that is currently very manual, though looking forward to automation. So what is scaling up is the hiring, but the process is being scaled out like the process of hematopoietic stem cell transplantation has been scaled out. In the autologous CAR-T setting, we have demonstrated durable responses, but relatively high cost of goods at present. In the allogeneic CAR-T setting, the cost of goods per dose is lower, but the long-term durable responses in absence of transitioning to a definitive therapy such as stem cell transplant are lacking. I don’t see it as ‘auto’ that has to be scaled out vs. ‘allo’ that can be scaled up, but ‘auto’ plus ‘allo’ for the reason that there are many patient from whom adequate autologous T cells cannot be generated.
Dr. Murthy: Allogenic therapies, which comprise off-the-shelf cell therapies, will solve a lot of the challenges that we face with autologous cell therapies, especially with respect to manufacturing. But those therapies are still several years away. We are likely to remain in the scale-out domain for a while with autologous therapies. Within the scale-out framework, I expect that we will see innovations in advanced analytics, decentralized manufacturing, and other areas. However, there are other issues of scale to consider within the scale-out framework, for example, ensuring reliable supplies of viral vector in large quantities. Here, the challenge is scaling up the production of a material that is essential for a scaled-out cell therapy (i.e., CAR-T). Thus, scale up versus scaleout is not necessarily as clear-cut of an issue as it might seem.
Dr. Sourdive: The players in the autologous field are really scaling out. It is a whole game of logistics, of centralized versus decentralized manufacturing. It is essentially also a race against time. How do you make sure that the cells that are drawn from a patient can be infused back into the patient post-processing in a limited amount of time with some appropriate control? In allogeneic settings, we have the opportunity to also scale up and be able to start with more material and grow larger-size batches. We have the capacity to serve the needs of many patients with material from single runs (already yielding hundreds of doses with today’s early processes). We think the allogeneic space has the opportunity to go for bigger scale up and not just scaling out; and that is clearly the trend that we see today.
GEN: What is one thing that researchers can do to ensure scalability for their cell-based therapy operations?
Dr. Adams: As with any process, to truly improve scalability, one must have ever-increasing knowledge of the mechanisms and failure modes associated with the product and the process. Reducing the use of exotic media components or highly specialized pieces of equipment makes it easier and more cost effective to replicate production suites and reduce site to site variability.
Dr. Langer: Researchers should start process development early and engage with CMOs or academic centers with experience in cell therapies as soon as possible. They should also try to base their cell engineering approach on platforms that have a clinical-scale analog.
Dr. Eglen: Plan from the beginning with the endpoint in mind. Often, processes that work well on a research scale need to transfer to bioproduction and be part of the upstream bioprocessing phase. It’s therefore important for research platforms to be not only efficient and cost-effective, but also scalable to the needs of the clinic. In addition, in this phase they need to incorporate automation and online monitoring.
Dr. Levine: Why does it have to be one thing? We are looking at automating unit operations of the process or, potentially, the entire process. Right now, it is manual, and I make the analogy to the Model T assembly line. It takes a lot of human labor to produce these therapies. The more we can automate, the more quickly and more efficiently we can generate these products. But unlike the Model T and other automobiles, the raw material in an autologous setting is different.
A second point to mention is that there will be conversion to electronic systems of the batch records and documentation, a lot of which is currently manual, and electronic management of manufacturing execution systems. Our knowledge will increase as we obtain more data from more patients on what algorithms to put into these automated solutions for cell processing.
Dr. Murthy: Be open to automation and embrace such approaches early in research. This is difficult for most life sciences researchers because it is daunting, and understandably so. But any reduction in manual processing, dependence on operator skill, and number of steps will improve the scalability of the process—or the development of any given cell therapy. This kind of approach also helps the robustness and reproducibility of the underlying science, which has tremendous value.
Dr. Sourdive: What we do when we have people working on a new product candidate or even the next generation of products is to ask them early on to take into account future deployment in a GMP environment, i.e., incorporate the constraints of downstream development into their plans. They need to know the ‘do’s’ and ‘don'ts’ from day zero. For example, some reagents or specific manufacturing steps are just off the table. The goal is to have the team say ‘OK, we are going to work on something new, but the toolbox is first limited to what can make sense in a GMP environment.’ We will use an approach or a process, or design a product with that in mind.
One important point is you need to know your starting material. When coming from healthy cells, it is much better for scalability than material coming from patients who have, for example, undergone several rounds of chemotherapy or who are in an advanced disease state. Allogeneic cell therapy is the preferred scalability platform because there is availability of a therapeutic product for hundreds of patients.
Cell Therapies Scale More Efficiently with Dissolvable Microcarrier-Based Cell Culture
Hannah J. Gitschier and Jennifer L. Weber
Many stem cells, such as human mesenchymal stem cells (hMSCs), are readily expanded in dishes and smaller flasks, but these expansion methods for process optimization and scalability can present challenges for cell therapy companies trying to achieve sufficient dosage for clinical applications.
One solution comes in the form of microcarrier-based culture in spinner flasks and stirred-tank bioreactors, which can provide high-density stem cell culture and mitigate risk for variability and contamination associated with multiple batches of smaller vessels. Corning Dissolvable Microcarriers further advance the cell therapy manufacturing process by providing a solution for simplifying the downstream processing and separation steps affiliated with microcarrier culture. Composed of cross-linked polygalacturonic acid (PGA) polymer chains, the microcarriers can be efficiently dissolved during cell harvest steps with a standard cell culture harvest solution containing EDTA and pectinase. This results in an easier and more efficient process.
Further, the dissolvable microcarriers are available pre-coated with either a denatured collagen or Corning Synthemax™ II surface to support stem cell expansion, health, and functionality. Several in-house studies have demonstrated multi-passage expansion of hMSCs on the microcarriers in both serum-containing and serum-free media conditions, while maintaining normal karyotype and multipotency. These microcarriers offer the opportunity to overcome some of the challenges and disadvantages associated with traditional microcarrier culture while still providing high cell yields for many cell therapy applications.
Hannah J. Gitschier is production line manager, bioprocess, and Jennifer L. Weber is development associate, cell biology, at Corning Life Sciences.
Corning® Dissolvable Microcarriers were designed to provide a solution for simplifying the downstream processing and separation steps affiliated with microcarrier culture.

