
April 1, 2016 (Vol. 36, No. 7)
DeeAnn Visk Ph.D. Founder and Principal Writer DeeAnn Visk Consulting
Just Because a G-Protein Coupled Receptor Has Resisted Characterization Doesn’t Mean It Can’t Be Sighted—or Hunted
For drug developers, G-protein coupled receptors (GPCRs) are dearly sought trophies. Many GPCRs have already been “bagged”—about 30% of all prescription drugs target a GPCR—but GPCR hunts continue, even though the quarry that remains is, naturally, more elusive.
Genomic studies reveal about 800 putative individual genes that are classified as GPCRs, roughly half of which are thought to be olfactory receptors. Of the other half, approximately 100 have no known function or ligand. These are called “orphan” GPCRs. Flushing them out is called “de-orphanization.” Almost as elusive as orphan GPCRs are poorly characterized GPCRs. Even GPCRs that have been de-orphanized may remain functionally and pharmacologically indistinct. There are about 200 of these hard-to-discern and hence not-quite-targetable GPCRs.
Although GPCRs may seem to be strange beasts, all but impossible to corner and hence resistant to characterization, it would be a mistake to conclude that GPCR hunts are impractical or even comparable to unicorn hunts. There is nothing fanciful about them. All they require is the patient application of improved experimental techniques.
Continued improvement in stabilization and crystallization of GPCRs permits the solution of their structures via X-ray crystallography. Deeper understanding of functional pathways activated by GPRCs allows better control of their signaling. Allosteric binding away from the ligand binding site permits finer and novel control of the signal produced by GPCRs. Also, in silico techniques make a powerful combination in the hunt for drug candidates.
The diversity of experimental and analytical techniques is complemented by a diversity of participants in GPCR hunting parties. With collaborations within academica and between academia and industry, multiple approaches are contributing to the pursuit and pharacological capture of GPCRs.
Against the Wall, Around the Bend
Several characteristics of GPCRs work in their favor as drug targets. As a transmembrane protein, GPCRs bind small molecules that are outside the cell, and so, drug candidates meant to bind with GPCRs do not have to cross the cell membrane to effect change. Aiming at GPCR targets means that drug developers need not consider the movement of compounds across the cell membrane.
Christian Felder, Ph.D., a research fellow in neuroscience discovery research at Eli Lilly and Company, sees GPCRs as interesting targets for mood disorders, mental health problems, and pain. GPCRs are cell surface proteins that bind neurotransmitters, and are therefore great targets for these maladies.
Studying the structure of GPCRs is challenging. “GPCRs experience a lot of wobble or movement within the cell membrane,” explains Dr. Felder. “This flexibility makes GPCRs difficult to crystalize. ” New technological advances, however, offer better methods to solving this problem, including protein engineering and lipidic cubic phase methodology.
For example, replacing the third transmembrane loop of a GPCR with a more stable amino acid sequence and then stabilizing the receptor in a type of lipid droplet is one way to help make GPCR receptors more amenable to crystallization. From there, traditional methods such as X-ray crystallographic diffraction can be used to solve the physical structures of GPCRs.
Dr. Felder is also interested in solutions to the selectivity problem. The root of this problem is that GPCRs are very homologous to one another. Most drugs cross-react with other similar receptors instead of only the specific GPCR that is desired.
A new approach to increase selectivity is to target allosteric binding sites or areas outside the neurotransmitter binding pocket for the modification of the action of GPCRs. When these allosteric sites are bound by a small molecule, the signal generated by the endogenous neurotransmitter can be modified. Screening assays can be designed to find compounds that interact with the GPCRs outside of the canonical ligand binding site.
“These aptly named allosteric modifiers preserve receptor fidelity of the time and location of the GPCR action,” says Dr. Felder. “This eliminates or greatly reduces off-target effects of traditional drugs.”
Positive allosteric modulators (PAMs) and negative allosteric modulators (NAMs) are a hot topic in drug discovery and development. These compounds can potentially allow the modulation of specific signaling pathways mediated by GPCRs. PAMs and NAMs can attenuate the signal in a positive feedback or negative feedback fashion.
Another interesting development in the field of GPCRs is the use of molecularly modified receptors that are activated solely by a synthetic ligand—a designer receptor exclusively activated by designer drugs (DREADD). This tool offers a unique option for studying the effect of GPCR downstream signaling.
First, the GPCR is mutated to respond only to a nonnative or synthetic ligand. Next, this construct is inserted into a mouse where expression of the DREADD can be spatially and temporally regulated. Then, when the modified mouse is given the synthetic ligand, information on the physiological function of that particular GPCR-regulated signaling can be determined in specific brain regions. Currently, DREADDs are a chemogenomic method employed by many laboratories as a more physiological technique to control GPCR-mediated signaling in a variety of cell types and locations.
“DREADDs are an excellent approach to gain knowledge on the physiological function of under-characterized GPCRs,” declares Dr. Felder. “The pathway inside the cell remains the same. It is the outside portion that is modified to respond to a nonnative ligand. Artificially stimulating the pathway by administering the synthetic ligand leaves all the downstream cascade effectors in place, allowing much insight into the endogenous function of the GPCR.”
To increase the success of finding a good therapeutic target, Eli Lilly likes to ask two key questions:
- Is the target related to the disease?
- Can modifying the GPCR target lead to a safe and effective therapy for the patient?
“One strength of asking these questions is to ‘de-risk’ the investment in drug discovery and development,” comments Dr. Felder. “Eli Lilly wants to deepen the understanding of GPCRs to lessen the risk involved in bringing a new medication to market. This is done in part by looking for key collaborations with academia as well as the strategic use of technological advances.”
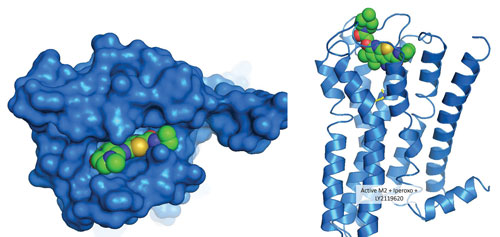
Researchers at Eli Lilly and Company have undertaken the exploration of probe dependence and the molecular mechanisms by which allosteric molecules may act upon a GPCR receptor and its signaling properties. Left: Surface representation of the muscarinic M2 receptor (a GPCR) with the positive allosteric modulator (PAM), LY2119620, bound in the outer vestibule. Right: Ribbon model of the muscarinic M2 receptor with the PAM, LY2119620, bound in the outer vestibule (space-filling model) and muscarinic agonist, iperoxo, simultaneously bound in the deeper orthosteric binding pocket (stick model). [© Copyright Eli Lilly and Company. All rights reserved. Used with permission.]
More Cloistered Settings
An example of the industry and academic collaboration can be found in the laboratory of Karen O’Malley, Ph.D., a professor of neuroscience at Washington University in St. Louis. She studies a GPCR named metabotropic glutamate receptor 5 (mGluR5). Her lab revealed the surprising finding that mGluR5 is expressed not only at the cell surface, but also in the nuclear membrane. The amount of mGluR5 found around the cell nucleus was three to five times higher than the amount found in the cell membrane.
To determine if theses seemingly out-of-place receptors actually propagated a signal, Dr. O’Malley collaborated with Eli Lilly, using drug candidates that did not have good profiles to be developed for human patients, but were extremely valuable in cell-based studies.
“Molecular genetics failed to remove all mGluR5 from the cell membrane,” says Dr. O’Malley. “So we isolated the function of mGluR5 pharmacologically. Eli Lilly provided a molecule from their library shown to selectively block mGluR5 on the cell surface, but not cross the cell membrane to the inside of the cell.
“Using this pharmacological blockade, we showed that the mGluR5 located on the cell membrane was indeed functional. We were very lucky to collaborate with Eli Lilly who kindly supplied an impermeable agonist and antagonist that allowed us to modulate mGluR5 on the cell membrane. Additionally, Eli Lilly supplied a transportable agonist and a permeable antagonist which we used to address the question of function for the internal mGlu5R.”
While looking for precisely which signaling pathways were activated, Dr. O’Malley found that mGluR5 on the cell surface activated the mTor-PI3K cascade. She also noticed that the intracellular mGluR5 activated only the MEK/ERK pathway.
Recently, the Dr. O’Malley’s laboratory collaborated with Terence Coderre, Ph.D., a professor of anesthesia research at McGill University, to show that blocking the intracellularly located mGluR5 receptors in the spinal cord of rats drastically reduced the pain experienced by the animals.
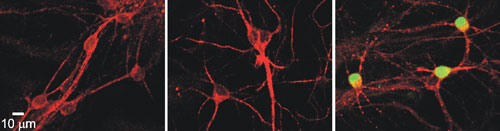
The laboratory of Karen O’Malley, Ph.D., at Washington University in St. Louis has focused on a GPCR called metabotropic glutamate receptor 5 (mGluR5). It is expressed not only at the cell surface, but also in the nuclear membrane. In this image, neurons stained for mGluR5 are shown in red, and a downstream signaling molecule, phosphorylated extracellular signal-regulated kinase (p-ERK), is shown in green. Left: Negative control neurons. Middle: Neurons treated with a nontransported, impermeable cell-surface agonist for mGluR5. Right: Confirmation of induction of mGluR5 by the activation of p-ERK after a five-minute treatment with a mGluR5 transportable agonist. This demonstrates that a GPCR can be activated within the cell, not just at the cell surface.
Stabilization of GPCRs
One company that has refined new approaches to obtaining the physical structure of GPCRs is Heptares. Tackling the issue of wiggly GPCRs, Heptares offers a unique platform to stabilize proteins allowing GPCRs to be removed from the membrane, crystalized, and studied with X-ray crystallography to determine their structure.
According to Dr. Cooke, the Heptares approach involves looking for unusual binding modes away from the binding site. An attractive mode is typically somewhere else on the protein—an allosteric binding site.
“Being unstable, GPCRs have presented quite a challenge in the past due to their notorious instability, especially when removed from the cell’s membrane,” states Rob Cooke, Ph.D., the head of biomolecular structure at Heptares. “By using Heptares’ proprietary techniques to stabilize GPCRs or other proteins of interest, users can gain the ability to crystallize heretofore uncrystallizable proteins.”
“Structures of several GPCRs with ligand classes A, B, and C have been solved,” asserts Dr. Cooke. “This atomic-level resolution paves the way for the design of drugs to bind to the protein. Through Heptares’ drug discovery technology, it is possible to use structure-based design to target GPCRs.”
Indeed, Heptares has an impressive list of customers, including AstraZeneca, MedImmune, MorphoSys, Pfizer, and Teva Pharmaceutical Industries. This is in addition to Heptares’ own internal pipeline.
One exciting application of Hepatares’ new STaR® (Stabilized Receptor) technology was the crystallization of the muscarinic M1 receptor. This technical achievement allowed Heptares to design a selective agonist against this receptor without cross-activation of the M2 or M3 receptors.
“In the past, drug candidates that targeted the M1 receptor never progressed beyond clinical trials due to the side-effect caused by this cross-reaction with the M2 and M3 receptors,” explains Dr. Cooke. “This is great news for all persons suffering (or who have loved ones suffering) from dementia or schizophrenia.”
Computer Simulations
The application of in silico screening of drug compound candidates is also booming. “With the new techniques available for solving the three-dimensional structures of GPCRs, laboratories can confidently move into the realm of computer simulations to find new ligands and binding sites for GPCRs,” says Brian K. Shoichet, Ph.D., a professor of chemistry at the University of California, San Francisco. “Modeling of GPCR structures has led to an explosion of research into using structure-based discovery and design.”
One example of the in silico approach can be found in the work done by the Shoichet laboratory and collaborators at the University of North Carolina. An orphan GPCR, GPR68, was known to be expressed in the hippocampus, but this receptor’s function remained a mystery.
A physical yeast-based screen showed the benzodiazepine drug, lorazepam, bound to an allosteric site on GPR68. “Exploiting our knowledge of GPCRs, we performed an in silico screen of 3.1 million compounds—real compounds that someone somewhere has offered to sell,” explains Dr. Shoichet. “Several potential hits from this screen were confirmed with functional assays.
“Ogerin, one potent modulator of GPR68, quelled the ability of wild-type mice to remember situations that were fear-inducing. Conversely, GPR68-knockout mice had no suppression of the ability to recall fear conditioning.”
This demonstrates a novel, powerful approach to finding new ligands for orphan GPCRs. The approach moved from computer simulations to in silico screens, to empirical screens, back to biological experiments in whole animals. Each of the approaches alone is strong, but combining them leads to the ability to resolve problems previously insoluble.
“Another exciting change is the paradigm used to think about the action of GPCRs,” declares Dr. Shoichet. “It is now known that GPCRs are not limited to stimulating just one downstream pathway. Instead, they can influence several pathways at the same time, possibly while introducing a bias toward one pathway or another.”
Downstream Effects
Downstream signaling is a built-in characteristic of GPCRs. Agonists potentiate GPCR signaling whereas antagonists attenuate it.
In general, GPCRs interact with three protein families in a conformationally sensitive manner—heterotrimeric G proteins, G protein-coupled receptor kinases (GRKs), and arrestins. Interaction with G proteins mediates intracellular signaling while interaction with GRKs and arrestins turns off G protein signaling, but stimulates GPCR endocytosis and arrestin-mediated signaling.
One good example of a GPCR whose response is regulated by G proteins, GRKs, and arrestins is the beta 2 adrenergic receptor. Functionally, this GPCR activates cAMP production and regulates physiological effects such as heart rate and airway smooth muscle relaxation.
“To address the issue of downstream signaling in the beta 2 adrenergic receptor, certain questions need to be asked,” says Jeff Benovic, Ph.D., professor and chair of biochemistry and molecular biology at Thomas Jefferson University. Specific questions posed by Dr. Benovic include the following:
- What is the conformation of the beta 2 adrenergic receptor when bound to agonist, when not signaling to the downstream arrestin pathway?
- Can we find small molecules that will stabilize this conformation that activates the downstream pathway of G protein and not arrestin?
“Answering these questions has potential for pharmacological approached to the treatment of airway and cardiovascular diseases,” asserts Dr. Benovic. “Being able to potentiate an established drug, or tweaking an existing one toward the G protein pathway, could alleviate side effects of beta-agonist medications used to treat asthma.”
One approach to studying conformational changes in beta 2 adrenergic receptor is to employ lipidated peptides called pepducins. These peptides bind to the GPCR, helping to stabilize different conformations, which in turn can be studied to see their effects on downstream signaling.
“Small molecules can also bind to allosteric sites and bias the signaling from beta 2 adrenergic receptors,” explains Dr. Benovic. “These compounds could be used as drugs to bias the signaling response of these receptors toward the G protein pathway (for treating asthma) or toward the arrestin pathway (for treating cardiovascular disease).”
“Using this theoretical framework leads to the design of screens that selectively promote G-protein coupling or arrestin coupling,” concludes Dr. Benovic. “We anticipate that utilizing this approach will yield more selective compounds that will have fewer side effects for many GPCRs.”
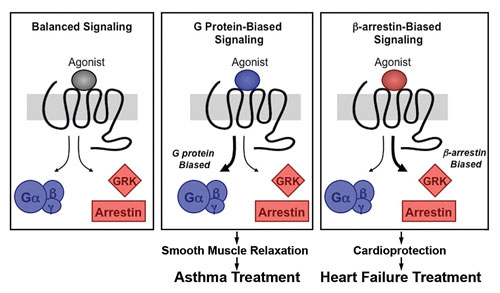
Schematic diagram of how signaling pathways can be biased toward a desired outcome, depending on the effect of an agonist binding to the GPCR (in this case the beta 2 adrenergic receptor). [Benovic lab/Thomas Jefferson University]





