
February 15, 2011 (Vol. 31, No. 4)
Steven Kain Ph.D. Director NuGEN Technologies
RNA-Seq Sample-Prep Reagents Aim to Help Analyze Previously Inaccessible Samples
The arrival of next-generation sequencing (NGS) technology has revolutionized the field of genomics and expanded the scope of scientific inquiry to sequence datasets from multiple genomes and transcriptomes. RNA-Seq is a specific technique enabled by next-generation sequencing and refers to direct sequencing of cDNAs to permit quantification of transcript levels from particular tissue or cell types.
Unlike hybridization-based methods such as microarrays, RNA-Seq also provides direct sequence information that can be used to compare single nucleotide variants, map transcription start sites, and detect novel transcript splicing. The digital precision and sensitivity of RNA-Seq is well suited to the analysis of low input samples, such as small populations of circulating tumor cells, cancer cells, and stem cells where a more detailed transcriptome map may reveal the true biology of such systems.
However, current technology for transcriptome sequencing requires a few hundred nanograms of total RNA (tens of thousands of cell equivalents). Moreover, many RNA-Seq protocols require additional enrichment steps to select for poly(A)+ RNA and/or to reduce the content of ribosomal RNA (rRNA) prior to NGS library construction. Procedures of this type require a higher amount of starting total RNA and also limit biological information derived from sequence analysis since often the whole tissue with a mixture of various cell types is used instead of pure cell populations.
The Ovation® RNA-Seq System from NuGEN provides a complete solution for the preparation of double-stranded cDNA for NGS library construction from inputs as low as 500 picograms of total RNA (~50−100 cell equivalents) allowing analysis of isolated cells of a particular cell type. The protocol does not require rRNA reduction or poly(A)+ selection, which may bias or provide an incomplete representation of the transcriptome.
The Ovation RNA-Seq FFPE System offers these same benefits and enables RNA-Seq analysis from formalin-fixed paraffin-embedded (FFPE) tissue, the most common source of archived clinical samples, especially in cancer studies. These RNA-Seq solutions have facilitated scientific discovery in diverse areas of research including gene-expression differences in bronchial airway epithelium associated with smoking and lung cancer, the characterization of the HIV genomes from clinical samples, and transcript profiling with RNA derived from FFPE samples.
Researchers at the Boston University School of Medicine lead by Avrum Spira are working toward the discovery and development of biomarkers for lung cancer and COPD that are detectable before the onset of clinical symptoms. These researchers are specifically interested in gene-expression changes that occur in the airway epithelial cells of tobacco smokers that may ultimately serve as biomarkers for disease risk in smokers.
The collection of sufficient total RNA for transcriptome analysis in such studies is limiting, as only a small number of the cells can be collected from the airway epithelium by bronchoscopy or using airway swabs. The differential gene-expression results shown in Figure 1 were generated using total RNA obtained from individuals who had never smoked and have no clinical indications of lung disease versus current smokers.
Total RNA was processed using the Ovation RNA-Seq System for analysis by NGS, or amplified and labeled for analysis on Affymetrix® Exon 1.0 ST microarrays. The results indicate that a greater number of differentially expressed genes are detected by NGS (region shown in green) owing to the greater sensitivity and dynamic range of this technique in comparison to microarray-based methods.
The RNA-Seq results provide a comprehensive and high-resolution view of the airway epithelial cell transcriptome and will provide insights into the molecular field of damage induced by smoking and the progression of tobacco-related lung disease. Moreover, each of the differentially expressed genes detected in the tobacco smokers are candidates for further development as lung cancer biomarkers.
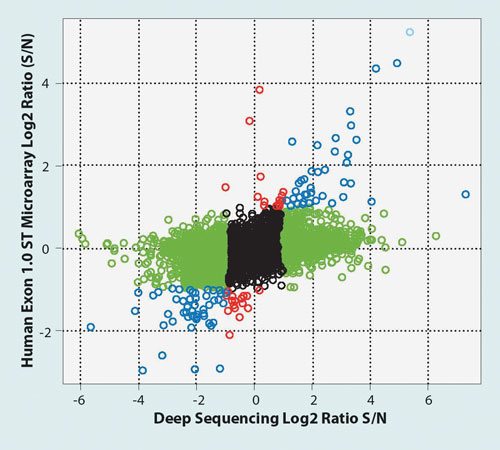
Figure 1. For the sequencing data, the expression of each gene was quantified in reads/million (RPM) based on the alignment of both ends of the paired-end sequencing experiment. For the microarray data, probes were summarized into gene-expression values using RMA and the Ensembl Gene CDF file. The Y-axis is log2 fold change by microarray and X-axis log2 RPM fold change by sequencing. Differential gene expression is defined as greater than a twofold difference between samples. The results indicate that 857 genes were differentially expressed in smokers versus never smokers (S/N) as detected by RNA-Seq, with 289 genes detected by microarray analysis.
Profiling the HIV Genome
It is estimated that 38 million people worldwide are infected with human immunodeficiency virus (HIV), with an additional 4.1 million people infected each year. With the growing HIV+ population it is important to understand how this virus mutates and develops drug resistance over time, which provides additional challenges in developing effective therapies.
Researchers at the University of California, Berkeley lead by Stephanie Willerth chose RNA-Seq to study HIV evolution in clinical isolates through a detailed analysis of viral sequences. However, there are several challenges in utilizing NGS to perform such studies. In particular, the amount of HIV RNA that can be obtained from clinical blood samples is low and does not provide enough material for traditional library construction protocols.
PCR amplification could not be used as PCR can introduce bias into a sample since only sequences that contain significant homology with the primers sequences will be amplified, which in turn affects the resulting analysis of diversity present in the HIV genomes.
In order to obtain sufficient quantities of material for NGS analysis, the researchers utilized the Ovation RNA-Seq System for low bias amplification of HIV genome samples to produce double-stranded cDNA, which was followed by library construction and sequencing on the Illumina Genome Analyzer platform.
The results produced high genome coverage using 36 base pair reads enabling accurate comparison of sequence differences. The researchers are currently developing metrics for evaluating HIV diversity and evolution and how these parameters change in response to long-term antiviral drug therapy. The methods developed in this study for low input RNA amplification can also be applied to other clinically relevant viruses.
FFPE tissue fixation has been the clinical sample archival storage method of choice for many years, in particular for cancer research and diagnostics. FFPE samples can be connected to patient data through healthcare, disease, and population registries and represent an important resource for retrospective studies of genomic-based disease.
While this preservation method renders the samples suitable for traditional cell and tissue characterization by microscopy, the FFPE samples are typically difficult to access for genomic analyses. RNA and DNA exposed to formalin fixation are often degraded and cross-linked, thereby making these samples refractory to the steps used in NGS analysis.
In order to access this important category of samples, NuGEN has developed the Ovation RNA-Seq FFPE System which is optimized for degraded RNA typically found in FFPE samples. Figure 2 shows an example of differential expression for a specific gene (TPM2) detected in a human colon tumor compared to normal adjacent tissue from FFPE and fresh frozen (FF) samples. The data show concordant several-fold reduction in the TPM2 transcript abundance in tumor in both FFPE and FF samples as indicated by the read distribution across exons. In addition, an alternative splicing event at Exon6 of the TPM2 gene is also seen in both the fresh frozen and FFPE samples (black arrows).
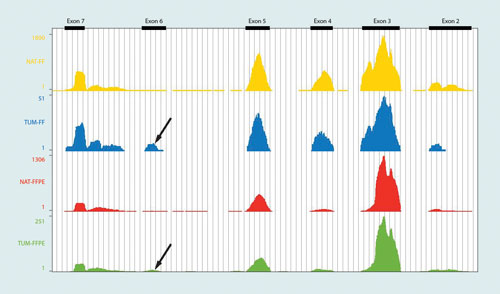
Figure 2. Differential expression and alternative splicing of tropomyosin 2 (TPM2). Data from UCSC genome browser illustrating read coverage of a portion of the TPM2 gene in human colon tumor vs. normal adjacent (NAT) samples. The data shows a concordant several-fold reduction in the TPM2 transcript abundance in tumor in both FFPE and fresh frozen samples (note the different Y-axis scales). An alternative splicing event at Exon6 of the TPM2 gene is also seen in both the fresh frozen and FFPE samples (black arrows).
Steven R. Kain, Ph.D. ([email protected]), is director of product management at NuGEN Technologies.





