
Jay Sandler
Marc Valer
Holly Hogrefe, Ph.D.
Site-directed mutagenesis (SDM) has been an important feature of the research landscape since its introduction in 1995. Continuing improvements to Pfu DNA polymerase have led to enhancements in the method, permitting protocols with higher fidelity, longer length-capability, multi-site targeting, and significantly faster workflows. The new trend in mutagenesis combines the advantages of random and site-directed methods (that is: the ability to survey large target regions with no prior structural knowledge while enabling user-specified changes, and with the potential to develop all possible variants) in order to create comprehensive, rationally designed mutant libraries. In this dynamic technological landscape, it is useful to survey how the features and benefits of the various methods have evolved and what this entails for the future of protein engineering.
Site-Directed Mutagenesis: Conventional Mutagenesis with Limited Scale
Conventional site-directed mutagenesis methods let you rapidly and efficiently create point mutations, amino acid substitutions, insertions, and deletions in virtually any double-stranded plasmid—useful for understanding protein-protein interactions and structure-function relationships. These methods are also commonly used for functional validation of genetic variants such as SNPs, codon optimization to increase protein expression, and for knocking-out undesired activity. However, because of the targeted approach, prior knowledge of the protein’s structure is required to achieve the desired mutation. In addition, researchers who wish to increase the scale of mutagenesis (beyond single-site mutations) may find these methods limited.
Multi-mutagenic methods such as QuikChange Lightning Multi, addresses some of these limitations by permitting rapid and efficient creation of point mutations in plasmid DNA at up to five sites simultaneously. While PCR-based methods are typically limited to two simultaneous mutational sites (each targeted with one of the PCR primers), by employing a unique multi-enzyme polymerase blend, multi-mutagenic methods are capable of incorporating up to five mutagenic oligos simultaneously in about three hours versus up to two weeks when using PCR-based methods. Such methods can be useful for identifying combinations of causal mutations that behave synergistically. Moreover, with a one-primer-per-site protocol, these methods can be used for codon saturation experiments, which employ degenerate codon (NNK; where N=AGCT; K=G/T) primers to explore the impact of all possible side chain replacements. However, like its site-directed counterparts, multi-mutagenic approaches also require some prior knowledge of the protein’s structure and its multi-site feature can be limited by a lack of knowledge of which positions affect function. Finally, while the Multi method can be scaled up to cover a complete domain, it could require tens to hundreds of reactions.
Random Mutagenesis: Surveying the Impact of Single-Base Changes across Genes or Gene Fragments
Random mutagenesis is a powerful tool for elucidating protein structure-function relationships and for modifying proteins to improve or alter their characteristics. For example, error-prone PCR is a random mutagenesis technique for generating amino acid substitutions in proteins by introducing mutations into a gene during PCR. Mutations are deliberately introduced through the use of error-prone DNA polymerases and/or reaction conditions. The mutated PCR products are then cloned into an expression vector and the resulting mutant library can be screened for changes in protein activity. Random mutagenesis allows researchers to identify beneficial mutations in the absence of structural information, or when such mutations are difficult to predict from protein structure.
Error-prone PCR is traditionally performed using Taq in the presence of Mn2+, which produces usable mutation frequencies, but impairs yield and target length (limited to hundreds of bps). Improved methods may feature robust polymerases that are engineered to reduce fidelity and blended to produce a uniform mutational spectrum across as many as 10,000 bps. Despite these improvements, a key limitation of error-prone PCR is library diversity. Amino acid changes that require two or three single-base replacements to occur sequentially in a codon will be poorly represented. It has been estimated that on average, only 7 amino acid replacements are possible for each codon.
Synthetic Biology: Combining the Advantages of Conventional Approaches to Provide Scalability
The gene synthesis approach, which has become more readily available in the last few years, allows researchers to synthesize any gene, multi-gene assembly, or gene variant at will. This approach has the benefit of creating only the desired variants, and avoids unwanted or nonproductive screening associated with degenerate-codon libraries (codon redundancy, stop codons, least-preferred codons, interference by wild-type). Custom mutant library synthesis is most effective for characterizing small numbers of variants (tens to hundreds), as the costs increase rapidly when building libraries to comprehensively uncover critical positions and identify improved variants across a complete domain. With custom mutant library synthesis, researchers focus on a limited number of specified mutant combinations, to avoid screening the large numbers required for representation when using combinatorial approaches with degenerate-codon oligos (i.e., 322=1,024 combinations of 2 codons, 32,768 combinations of 3 codons, etc.).
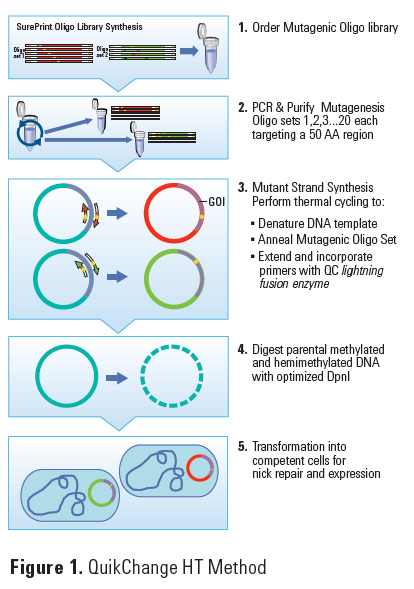
A new high-throughput site-directed mutagenesis method addresses the cost constraints of custom mutant library synthesis, while overcoming the screening burden posed by degenerate-codon oligos. The method combines high fidelity massively parallel oligo synthesis with the ease of use and efficiency of conventional site-directed mutagenesis. This new method enables Alanine Scanning (or single amino acid) and Codon Saturation Scanning at up to 10–20 sites (in one plasmid or different plasmids), where each target region or site spans up to 50 amino acids. Researchers can survey the impact of substitutions at up to 1,000 different amino acids, or test up to a hundred thousand combinations of key mutations across 20–50 contiguous amino acids. With this method, the incremental cost of a designed variant transformed into a competent cell is tens to hundreds times less than with synthetic gene variants. Building off of conventional site-directed and random methods, this new approach enables rapid resolution of structure-function questions, with the ability to create rational variant libraries of up to 120,000 recombinant mutants, making the elucidation of protein-protein interactions and structure-function relationships available at a truly large scale, accessible to more researchers.
For more on mutagenesis, be sure to check out “Enabling Fast, Efficient, and High Fidelity Site-Directed Mutagenesis through Continuous Innovation“.
Holly Hogrefe, Ph.D., is NGS R&D Director at Agilent Technologies.





