
January 1, 2010 (Vol. 30, No. 1)
Lee Pullan Ph.D.
Marc Storms Ph.D.
Cryo-EM Provides a Window on the Molecular Machinery of Biological Systems
Recent developments in transmission electron microscopy (TEM) have generated a renaissance in biological imaging, allowing researchers to visualize the 3-D structure of biological entities including viruses, protein complexes, and individual proteins at the nanometer scale. Equally important is the refinement of cryogenic sample preparation and sample-handling techniques that permits investigation of these structures in a fully hydrated state and in their native context.
Some of the most dramatic results have come in the field of virology, where new discoveries have revealed, not only the detailed structure of clinically significant viruses, but shedding a new light on mechanisms of replication, assembly, and cellular entry that suggest opportunities for new approaches.
Although TEMs routinely resolve individual atoms in material science applications, they face a number of difficult challenges in biological imaging, all related to the nature of biological samples. These challenges include practical requirements for samples that are compatible with the vacuum environment of the electron optical system and thin enough to transmit electrons and avoid ambiguity in the projected image caused by overlapping structures.
The most fundamental challenge comes from the inherently weak contrast of biological materials and their vulnerability to damage by the electron beam, which precludes the use of long exposures that could otherwise build image contrast and improve signal-to-noise ratios.
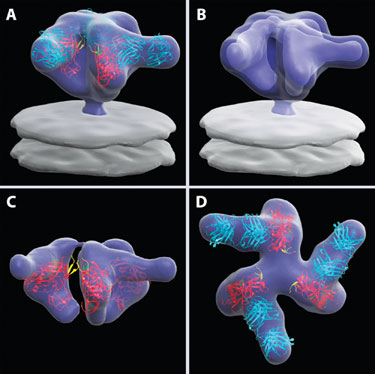
Figure 1. Averaged 3-D structure of the HIV-1 spike in complex with b12-Fab
Sample Prep
Cryo electron microscopy (cryo-EM) relies on a physical process known as vitrification, in which the temperature of the sample is lowered so quickly that water molecules do not have time to crystallize, instead forming vitreous ice, an amorphous, glass-like solid.
By avoiding the structural disruption caused by expansion during crystallization, cryo-EM preserves biomolecular structures in their natural, fully hydrated state. It literally freezes the sample at an instant in time and can be applied to molecular complexes, whole cells, and tissues, permitting observations of molecular components in association with their functional complements in their native context. The nearly instantaneous nature of the process allows time-resolved analysis of dynamic phenomena such as molecular flexibility, conformational changes, and intermolecular interactions.
High-contrast, high-resolution, low-noise images are difficult to obtain from biological samples because they do not typically contain features such as regular periodic structures or large variations in density or atomic number that generate strong contrast in a TEM; and they do not tolerate the high beam currents or extended exposure times that would build contrast and enhance signal-to-noise ratios. Low-dose imaging refers to a combination of techniques that seeks to address this problem by extracting the maximum information from the sample for each beam electron to which it is exposed.
Two different approaches to 3-D TEM analysis—single-particle analysis and tomographic analysis—have yielded important results. Single-particle analysis combines images from many separate but identical particles to construct a 3-D model.The images of arbitrarily oriented particles are first sorted into categories by similarity, with each category representing a similar orientation.
Images within each category are averaged to create a high-quality, low-noise composite image, then the composite images are combined in a 3-D model. Theoretically, the quality of the model can be improved almost arbitrarily by increasing the number of images.
Tomography computes the 3-D structure by recombining a series of 2-D images acquired over a range of perspectives as the sample rotates in known increments about an axis perpendicular to the beam direction. Its principle advantage is that it looks at a single instance of the imaged structure and can thus be used to evaluate variations among individual particles.
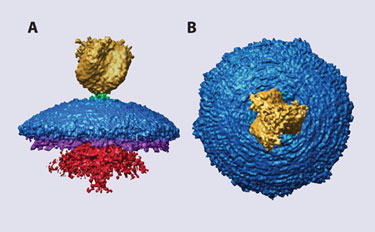
Figure 2. SARS corona virus merging cryo-EM and atomic resolution 3-D structures
Recent Research
The envelope glycoproteins (Env) of human and simian immunodeficiency viruses mediate virus binding to the cell surface receptor (CD4) on target cells to initiate infection (Figure 1). Using cryo-electron tomography combined with 3-D image classification and averaging, Sriram Subramaniam, Ph.D., head of the biophysics section in the laboratory of cell biology at the NCI and his colleagues have reported on the 3-D structure of Env.
By fitting known crystal structures into the density maps derived by electron tomography, they derived molecular models for the native Env, unbound and bound to the surface receptor. They demonstrated that binding results in a major reorganization of Env, leading to closer contact between the viral and target cell membranes.
The SARS coronavirus (SARS-CoV) spike is the largest known viral spike molecule (Figure 2). Daniel Beniac, Ph.D.’s group at the Public Health Agency of Canada are using cryo-EM and image processing (single-particle analysis) to investigate conformational changes that occur in the entire spike of intact virions when they bind to the viral receptor. They have shown that ACE2 binding results in structural changes that appear to be the initial step in viral membrane fusion. The SARS-CoV spike provides an ideal model system to study receptor binding and membrane fusion in the native state.
Investigators in the lab of Hong Zhou, Ph.D., professor of microbiology, immunology, and molecular genetics at UCLA, have reported on the 3-D structure of cytoplasmic polyhedrosis virus with 3.88 Å resolution using cryo-EM and single-particle analysis (Figure 3). This is reportedly the highest obtained resolution so far using cryo single-particle analysis. They confirmed the resolution of their analysis by identifying characteristic protein structures with well established dimensions such as the 5.8 Å pitch of an α helix and the 3.8 Å spacing along the polypeptide chains in an α sheet.
Recent developments in cryo electron microscopy—single-particle analysis and dual-axis tomography—have made it possible to investigate the 3-D structure of viral proteins and the complex interactions of these proteins with other macromolecular structures such as the molecular epitopes that are recognized by the immune system.
Cryo-EM has enabled important discoveries about the relationship of structure and function in the complex processes involved in viral replication, assembly, and infection. It has been shown to be a reliable and complementary adjunct to x-ray and NMR analysis. Unlike x-ray and NMR, which must look at large numbers artificially produced and purified proteins under conditions that are far from physiological, cryo-TEM offers the advantage of looking at the total functional entity in its natural context, thereby providing a unique and powerful window on the molecular machinery of biological systems.
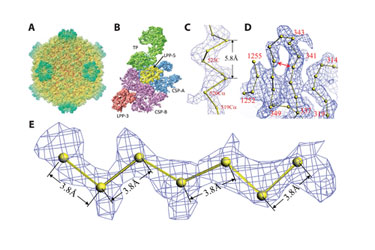
Figure 3. Structure of the CPV capsid
Lee Pullan, Ph.D. ([email protected]), is senior applications engineer, and Marc Storms, Ph.D. ([email protected]), is product marketing manager, structural biology solutions, at FEI. Web: www.fei.com.





