As a result of recent progress in DNA sequencing technologies, which has generated datasets of unprecedented complexity and advanced our understanding of health, disease, and development, whole-genome sequencing has emerged as a highly anticipated goal. However, routine sequencing of whole genomes is still a challenging milestone, particularly for clinical applications.
A more affordable approach, target enrichment, involves capturing genomic regions of interest prior to sequencing. One example is whole-exome sequencing, in which next-generation technologies sequence only the coding regions, which represent slightly over 1% of the human genome and are thought to harbor a large proportion of the variation associated with human disease.
“A few years ago, I would have thought that targeted sequencing would be influential for only a few years, and that it would be replaced, as sequencing costs come down, by whole-genome sequencing, but that has not happened yet,” says Chad Nusbaum, Ph.D., co-director of the genome sequencing and analysis program at the Broad Institute.
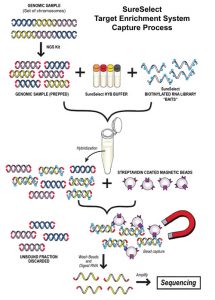
Dr. Nusbaum and colleagues developed an approach called Solution Hybrid Selection capture, in which biotinylated RNA capture bait probes are generated and mixed with a library of randomly sheared DNA fragments amplified from human genomic DNA that are modified with sequencing adaptors. Hybridized fragments are captured on streptavidin beads, and the DNA is sequenced on a next-generation platform.
This method facilitated the extensive sequencing of targeted genomic loci of interest. After optimizing and further developing this application, investigators from Dr. Nusbaum’s lab recently described the first automated, highly scalable application to perform Solution Hybrid Selection capture in a cost-effective and highly efficient manner.
Several target-enrichment approaches have emerged in recent years. A factor that has catalyzed the expansion of this field is that as long as the costs remain significantly lower than the costs of sequencing whole genomes, more samples can be analyzed. “Since human genetics is all about statistics, and statistics is driven by the number of samples, targeted sequencing remains a significantly growing area,” says Dr. Nusbaum.

Among target-enrichment platforms, exome sequencing has received increasing attention. To a great extent, this can be explained by our better understanding of the direct biological implications of sequence variations within exons. “The remaining 97–98 percent of the genome, while it cannot be ignored and we need to learn more about it, is still difficult to understand in terms of biological and clinical implications,” emphasizes Dr. Nusbaum.
“We were interested in examining quantitative trait loci, and after localizing a specific genomic region identified by linkage analysis, we adopted the emerging technology of targeted resequencing,” says Jeremy B.M. Jowett, Ph.D., head of genomics and systems biology at the Baker IDI Heart and Diabetes Institute from Melbourne.
By using the Agilent Technologies SureSelect target-enrichment system, Dr. Jowett and colleagues reported the possibility to combine index barcode multiplexing with solution-based target enrichment. The authors genotyped a 3.3 Mb region on the X chromosome in five individuals, and illustrated the strength of this approach in detecting most SNPs within the region, with a concomitant decrease in the time and costs involved.
“As this application becomes more mature and more robust, it will also find its way into the clinic,” says Dr. Jowett.
The ultimate goal, particularly when looking for mutations associated with complex diseases, is to perform whole-genome sequencing to capture all the genetic variation, quantitate the effect on disease risk, and identify regions and variants associated with specific conditions. Nevertheless, at least at present, this approach is challenging. “It might be too expensive to sequence whole genomes in hundreds of people, but it is possible, and within the budget of many labs, to conduct exome sequencing,” says Dr. Jowett.
The choice between whole-genome sequencing and target-enrichment depends on the specific application. Whole-genome sequencing could be the method of choice for research applications, but in the clinic, it might not be the preferred option, as it is more efficient to identify and focus on the genomic regions that are important for specific conditions and subsequently perform target enrichment.
“It may also be possible that the paradigm will be to initially focus on specific genomic regions containing genes associated with a disease, and this could involve a dozen or so different genomic regions, and if this would not reveal anything, there may be an additional step toward whole-genome sequencing.”
Exome Sequences
In many research articles, investigators perform exome sequencing to identify genes that are mutated in specific medical conditions. “We pursued a different approach, and in a cohort of people who had their genomes sequenced for completely unrelated reasons, we wanted to find out how many individuals have variations in a specific recessive disease gene,” says Leslie G. Biesecker, M.D., chief and senior investigator at the genetic disease research branch at the NIH.
Dr. Biesecker and colleagues relied on the ClinSeq cohort, a pilot project that uses whole-genome sequencing to investigate the genetic basis of health, disease, and drug response, and currently enrolls close to 1,000 individuals, in which the whole exome sequences are available for approximately 600.
The authors looked for genes involved in combined malonic and methylmalonic aciduria, a recessive Mendelian disorder, and found mutations in ACSF3, marking the first time a human disorder was causally linked to variations in an acyl-CoA synthase family member.
“We examined the exome sequences to find out how many individuals in the cohort have variations in that gene,” explains Dr. Biesecker. This approach not only identified recessive carriers of the mutation but also unveiled an example when two mutations were present in the gene.
“We previously thought that this is a severe disease with childhood onset, and finding in this adult cohort a patient with two mutations in the gene was quite unexpected.” Additional metabolic testing on the patient confirmed the disease, and revealed that this disease is not only a childhood onset severe metabolic disorder, but may also present as an adult onset mild condition that masquerades as a neurodegenerative disease later on life.
“This tells us that a genome-based approach may identify individuals with phenotypes that we do not even know that we should be looking for, and we can identify them that way.”
For certain biological questions, sequencing several exomes is more powerful than generating one whole-genome sequence, and this concept is illustrated by another recent finding from Dr. Biesecker’s lab. Dr. Biesecker and colleagues recently examined patients with the Proteus syndrome, a rare developmental condition with multisystem involvement and broad clinical variability, characterized by severe malformations and overgrowth of multiple tissues.
Exome sequencing from affected and unaffected tissues unveiled somatic mutations in AKT1 in 26 of 29 individuals with this condition, only in the affected tissues. “We also conducted high coverage whole-genome shotgun sequencing in a patient with this condition, and on a paired set of unaffected and affected tissues, we did not see the alteration,” reveals Dr. Biesecker.
“This is a clear example when sometimes lower costs of an exome, and the ability to interrogate more samples is more effective, whereas the whole genome, even though it provides more coverage, could miss things.”
Barcoded Samples
“We started off at a time when in solution target enrichment was not yet available, and relied, initially, on array-based enrichment,” says Edwin Cuppen, Ph.D., professor of genome biology and human genetics at the Hubrecht Institute, KNAW.
Array-based capture is technically laborious, and this is one of the reasons why the field has increasingly moved toward solution-based enrichment. “The weakest point from a diagnostic perspective is when certain bases are not covered well, and for this reason, we found microarrays to be much better for enrichment, because they provide higher and more even coverage,” explains Dr. Cuppen.
In an attempt to scale microarray-based enrichment, Dr. Cuppen and collaborators barcoded the index samples prior to sequencing and enrichment. “Instead of several enrichments in parallel, we wanted to see if we can mix the samples together and perform one enrichment,” explains Dr. Cuppen, who with his group illustrated the feasibility and the strength of this approach.
Target enrichment of individually multiplexed barcoded samples was performed with Life Technologies’ Applied Biosystems SOLiD-based next-generation sequencing technology in a single assay. This approach enabled coverage of the complete coding sequence of 770 genes from a 1.4 Mb genomic region, and identified new variants with over 96% sensitivity, while the false positive rate remained lower than one in eight Mb.
More recently, Dr. Cuppen and colleagues also illustrated the feasibility of this strategy for solution-based target enrichment, and successfully used this highly flexible and scalable setup for a wide range of multiplexing applications.
Hybridization Platform
In 2007, Richard A. Gibbs, Ph.D., professor and director of the human genome sequencing center at Baylor College of Medicine, together with colleagues from Roche NimbleGen published one of the first reports on the use of solid-based hybridization based enrichment of human genomic regions by programmable custom high-density oligonucleotide microarrays. “We have incrementally improved our reagents since then,” says Dr. Gibbs.
Most recently, Dr. Gibbs and colleagues described a liquid-phase hybridization platform that uses biotinylated oligonucleotide probes, and introduced additional design changes to include more genes than the narrow consensus coding DNA sequence (CCDS) set, which frequently guides the design of custom probes but excludes many computationally predicted or actual coding exons present in other databases.
To expand the regions examined during target enrichment, the investigators included two new reagents. The first one, VCR-set, includes microRNAs, Vega (the Vertebrate Genome Annotation Database), CCDS, and the RefSeq databases. The second capture design reagent, REC-set, additionally includes regulomes, exons, and conserved elements. By using these reagents, Dr. Gibbs and his team conducted the first genome-wide targeted capture analysis of a diverse set of biologically relevant genomic elements, and revealed decreased capture of variants located outside the CCDS regions as compared to the CCDS exome.
The results also showed that conserved untranslated regions, which are approximately 30% GC rich, and regulatory regions, which are approximately 70% GC rich, had approximately half of the depth sequence coverage following the capture procedure when compared to the CCDS regions, demonstrating the need to increase coverage in genomic regions that are different from CCDS.
At the biological end, Dr. Gibbs and colleagues are applying these advances toward the discovery of disease alleles and the study of rare genetic variants. “Having illustrated the robustness of this approach in the research arena, we are on the verge of developing this into a diagnostic test in the clinical arena.”
Solution-based hybridization approaches are often more convenient than solid-phase arrays, and offer the additional advantage of being easier to multiplex. “An important improvement from our point of view, particularly as we have been using the Illumina technology for next-generation sequencing, is that we are using TruSeq, the new library preparation system that Illumina has developed,” says Ann-Christine Syvänen, Ph.D., professor of molecular medicine at Uppsala University.
While many sequencing efforts focus on capturing exomes, a significant amount of genetic variation occurs outside protein-coding regions. “It would be important to also analyze other genomic regions, and the new enrichment probes from Illumina contain some extra sequences in regulatory regions very close to genes, which add information content in addition to multiplexing.”
These short regions that flank the genes allow the detection of regulatory variants located in their vicinity. In addition, genomic variation may also come from gene regulatory elements located further off from open-reading frames. This source of variation is also functional, but it will be missed by standard exome arrays.
Target enrichment and whole-genome sequencing emerge as two equally important strategies, and each of them is best powered to address specific biological questions. As these approaches are incrementally improved, optimized, and validated in research settings, they promise to materialize into exciting diagnostic and therapeutic applications and to provide powerful tools to interrogate other biological questions.





