There’s a new RNA-seq in town. Although the old RNA-seq administered a certain rough justice, profiling RNA species well enough to support a relatively crude conception of the transcriptome, the new RNA-seq is more refined. Yet, like the old RNA-seq, the new RNA-seq is quick on the draw. In fact, the new RNA-seq is compatible with the newest high-throughput technologies.
But the new RNA-seq is also bioinformatically up to date, capable of single-cell and single-nucleus analyses, and alert to all sorts of transcripts—even shifty splice variants, unstable pre-mRNA species, and elusive post-translational forms.
The new RNA-seq, or RNA sequencing, is what researchers and translational scientists need to develop a more sophisticated conception of the transcriptome. RNA-seq is characterizing previously unknown cell types and recognizing intermediate developmental states. It is uncovering epigenetic modifications that culminate in cancer. And it is relating the maturation and decay of RNA species to processes that underlie health and disease.
Getting a Bead on Single Nuclei
At the Broad Institute of MIT and Harvard, scientists have developed a droplet microfluidic and DNA-barcoding technique called DroNc-seq. Designed for the analysis of single nuclei, DroNc-seq combines single-nucleus RNA sequencing (sNuc-seq) with Drop-seq, a technology first reported in 2015.1 In Drop-seq, messenger RNA transcripts are profiled simultaneously from thousands of individual cells by associating them with a unique barcode for each cell and partitioning them into nanoliter-sized aqueous droplets.
“When we developed DroNc-seq, we built on a huge knowledge base and transferred the droplet-microfluidics approach to the nuclei technology to achieve the next level of scale,” says Naomi Habib, Ph.D., a postdoctoral fellow in laboratories led by Aviv Regev, Ph.D., and Feng Zhang, Ph.D., at the Broad Institute.
Dr. Habib participated in a proof-of-concept study that used DroNc-seq to interrogate gene expression in over 39,000 nuclei from mouse and human brain samples (Figure 1).2
“We found expression patterns that correlated very well with previous expression patterns that investigators found using more low-throughput methods,” asserts Dr. Habib.
In the study, DroNc-seq was able to cluster neurons of the same class originating from different anatomical regions of the brain, and it allowed different glial cell types to be differentiated from one another, despite the lower nuclear RNA content and the lower number of detected genes. DroNc-seq also captured more discrete differences between cells. For example, it distinguished between different types of GABAergic neurons that expressed different gene signatures.
“As part of our work, we also presented proof of concept that we can apply our strategy to frozen, archived brain samples,” notes Dr. Habib. On frozen 3–5-year-old postmortem archival human tissue, and despite variations in the sample quality, DroNc-seq generated high-quality libraries from both neurons and glial cells, including some rare cell types. “This means that we can utilize frozen tissue banks to explore samples at the single-cell level,” suggests Dr. Habib.
To accommodate the lower amount of RNA in nuclei compared to cells, the investigators found that they had to modify the Drop-seq technique. For example, the investigators changed the microfluidic device to generate smaller droplets and increase the efficiency of RNA capture.
The technical differences between the lysing of nuclei and the lysing of cells—differences related to the distinct properties of these membrane types—opened additional challenges. “This required knowledge from both the molecular biology side and the microfluidic side to be combined, to generate an optimal setup,” recalls Dr. Habib.
A very different type of technological challenge is that even though RNA can provide ample information about single cells, understanding the various functions of a cell requires several additional types of data to be extracted. “We would like to be able to obtain, from the same cell, knowledge about the epigenetic state, the chromatin state, and proteins, along with other types of information, such as [the cell’s] special position and its neighboring cells,” declares Dr. Habib. “This is a major and fascinating challenge that people are already working on.”

Figure 1. At the Broad Institute of MIT and Harvard, researchers Tyler Burks, Ph.D., and Naomi Habib, Ph.D., participated in a study that demonstrated the usefulness of DroNc-seq, a technology that combines single-nucleus RNA sequencing (sNuc-seq) and droplet-generating microfluidics. DroNc-seq, which puts sNuc-sec on a massively parallel basis, allowed Broad scientists to profile more than 39,000 nuclei from mouse and human archived brain samples to demonstrate sensitive, efficient, and unbiased classification of cell types. According to the scientists, DroNc-seq paves the way for systematic charting of cell atlases.
Oncofetal Epigenetic Control
“We need the most comprehensive and the most accurate and informative approach to look at gene expression, and RNA-seq provides that for many cancer types,” says Gary S. Stein, Ph.D., professor and chair of the department of biochemistry at the University of Vermont Larner College of Medicine, and director of the University of Vermont Cancer Center.
Investigators in Dr. Stein’s lab use RNA-seq for applications such as gene-expression analysis. This particular application helps Dr. Stein’s lab assess developmental processes, malignant transformations, and therapeutic responses.
“RNA-seq not only provides an opportunity to look at the mRNA that encodes proteins, it also provides informative about the full spectrum of transcripts, including noncoding RNA species such as microRNA (miRNA), transfer RNA (tRNA), toxic small RNA (tsRNA), and long noncoding RNA (lncRNA)—and we look at all of that,” explains Dr. Stein.
In the 1980s, Dr. Stein and colleagues cloned the human histone genes for the first time, and for the past few decades, these investigators have devoted much of their work to interrogating the genetic and epigenetic mechanisms of cell-cycle control that influence development and malignancy. One of the recent advances in Dr. Stein’s laboratory has been the finding that the early stages of some cancers recapitulate certain mitosis-specific bivalent histone modifications seen in pluripotent stem cells (PSCs).
Bivalent chromatin marks, which are defined as the presence of both the activating trimethylated histone 3 lysine 4 and the repressive trimethylated histone 3 lysine 27 modifications at gene promoters, were described over a decade ago in PSCs. Bivalent chromatin landscapes help establish the cancer state or the pluripotent fate in cells, notes Dr. Stein, through a process he calls “oncofetal epigenetic control.”
Studying this process could improve the mechanistic understanding of malignancies and lead to the development of novel diagnostic and therapeutic interventions.
In a recent effort to identify histone modifications that occur as part of differentiation programs in mesenchymal stromal cells, Dr. Stein and colleagues combined the study of several post-translational histone marks at a genome-wide level with RNA-seq.3 This approach demonstrated the complexity and the dynamics of the gene expression programs that characterize osteoblast differentiation from mesenchymal stromal cells.
Unlike other studies of bivalent histone modification, the new study found no active mechanism of gene repression in osteoblastogenesis. Instead, the new study suggests that epigenetic gene repression results from the loss of activation marks.
“It will be important to see additional developments in bioinformatics of how to interrogate RNA-seq data,” concludes Dr. Stein. “While there are some very good approaches right now, they are going to get even better.”
Splice Variant Bioinformatic Analysis
“As a bioinformatics team, we help generate novel scientific insights either through reanalysis of public datasets from sequencing archives or through our scientific collaborations with researchers all over the world,” says Jean-Noel Billaud, Ph.D., senior principal scientist at Qiagen.
Using their bioinformatics software portfolio, scientists at Qiagen extract expression profiles from RNA-seq sequencing reads and send them for biological exploration to their flagship software, Ingenuity® Pathway Analysis (IPA®).
IPA, a software for analyzing and interpreting omics data, is widely used by academic institutions, government laboratories, and pharmaceutical companies. “We use IPA, along with data derived from RNA-seq, microarray profiling, metabolomics, or proteomics, to help us understand and interpret the differential-expression pattern observed between two conditions,” notes Dr. Billaud.
The powerful analysis and search tools that are part of the IPA platform can, in the context of biological systems, identify the transcriptional programs that lead to a specific gene-expression pattern and highlight the biological processes involved.
IPA relies on the manually curated Qiagen Knowledge Base, which has been in continuous development for more than 15 years and helps place findings into their biological context. In combination with powerful algorithms, IPA provides advanced data analysis and interpretation capabilities and helps generate new hypotheses that can be tested and validated experimentally.
At a recent workshop in San Francisco, CA, Qiagen showed how IPA helped identify biological signatures that could distinguish melanoma patients who would respond to immune checkpoint inhibitor therapy from those who would develop resistance (Figure 2).
“A key benefit of IPA is the understanding of transcript-level biology as opposed to only the gene-level information,” explains Stuart Tugendreich, Ph.D., global product manager of IPA. For years, one of the challenges in data analysis has been the difficulty in analyzing and understanding the biological significance of the hundreds or thousands of different splicing isoforms that are generated by RNA-seq.
“IPA functionalities now enable researchers to start looking at those splice variants and to begin understanding their biology,” adds Dr. Tugendreich.
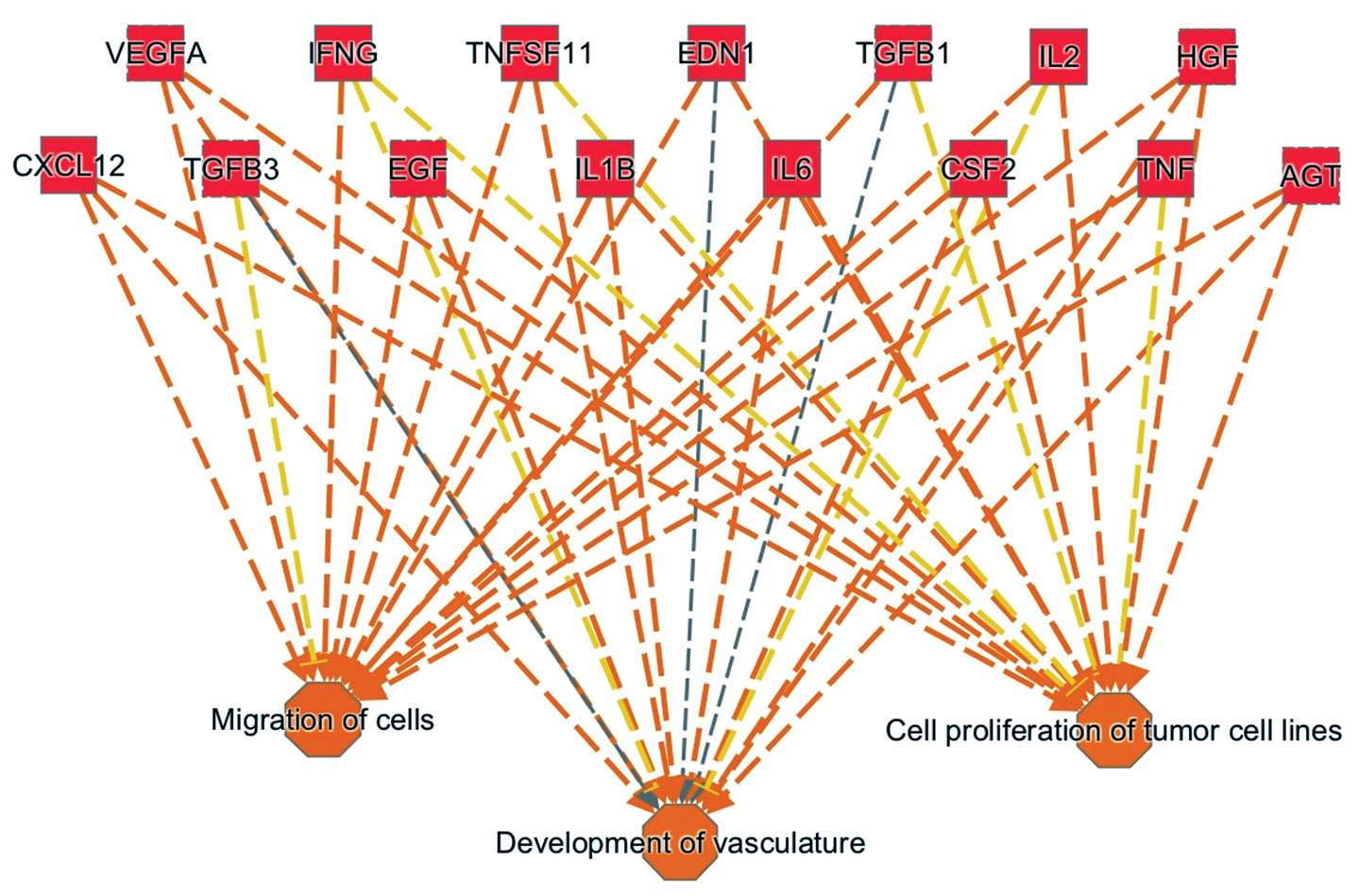
Figure 2. This pathway analysis diagram highlights the cytokines and growth factors involved in driving the tumor progression observed in metastatic melanoma patients who exhibit innate resistance to anti-PD1 treatment. Overall, the diagram presents an upstream regulator signature. It was generated by combining technologies from Ingenuity Systems and OmicSoft (both Qiagen companies). For example, Ingenuity Pathway Analysis (IPA), which incorporates a feature called Analysis Match, allowed a search of similar (or dissimilar) activation events across thousands of preanalyzed datasets. This search yielded the upstream regulator signature shown here, a signature that also emerges from other cancer datasets and may serve as a biomarker for predisposition to tumor progression. The underlying dataset is at the Gene Expression Omnibu (GSE78220).
Learning about RNA Decay
In the laboratory of Hamed S. Najafabadi, Ph.D., assistant professor in the department of human genetics at McGill University, several projects are examining RNA stability and dynamics in neurodegenerative conditions and cancer. “The main problem that we had during our work was that we could not look at the rate of RNA decay directly,” says Dr. Najafabadi. “We had to look at the abundance of RNA instead.”
“We have been trying to figure out how RNA stability changes across tissues or over time,” he continues. “Measuring RNA decay rates directly would give us more statistical power and a more direct way to look at the different factors that regulate RNA stability.”
Historically, looking at the decay of mRNA has been experimentally challenging. In fact, according to Dr. Najafabadi, anticipating the course of RNA decay can be like guessing the trajectory of a pitched baseball when your only clue is a snapshot of the pitcher. If you look at the ball, and nothing else, you will guess poorly. “But that is what we have done for a long time,” insists Dr. Najafabadi. “We have focused only on the abundance of the mature mRNA.”
Looking at both the hand and the ball, however, can provide insights into the ball’s trajectory. “This is what we started to do,” maintains Dr. Najafabadi. “By focusing on mature mRNA and pre-mRNA, we found a way to look at the trajectory over time. We can begin to understand how mRNA concentration changes over time and how mRNA degrades.”
In previous in silico research that separated intronic and exonic RNA-seq reads to dissociate the transcriptional and post-transcriptional contributions to gene expression, it was proposed that exonic reads reflect the steady-state mRNA abundance, whereas intronic changes reflect transcriptional dynamics. “We leveraged this observation and came up with a direct measure of the mRNA decay rate,” explains Dr. Najafabadi. By decoupling mRNA transcription changes and mRNA decay rates, Dr. Najafabadi and colleagues modeled the kinetics of mRNA metabolism and used a computational approach to estimate the differential mRNA decay rate.
Using a diverse panel of 20 different human tissues, Dr. Najafabadi and colleagues looked at the mRNA stability landscape in different cell types.4 “The mRNA stability landscape in the brain was quite different from what we saw in other human tissues,” reports Dr. Najafabadi.
This observation pointed toward the contribution of post-transcriptional programs to shaping the brain transcriptome, reinforcing similar observations that were made earlier using several different experimental approaches.
From RNA-seq measurements in the brain tissue, Dr. Najafabadi and colleagues identified four microRNAs and two RNA-binding proteins that were critical determinants of mRNA stability. One protein found during this work, RBFOX1, is involved in regulating the stability of mRNAs that encode synaptic transmission proteins.
“Loss of synaptic function is a major change in Alzheimer’s disease,” notes Dr. Najafabadi. “Accordingly, we surmised that in Alzheimer’s disease, there is a good chance that RBFOX1 is involved.”
Dr. Najafabadi and colleagues found that the mRNAs encoding synaptic transmission proteins degraded much more rapidly in the brains of individuals with Alzheimer’s disease. These investigators also determined that RBFOX1 inhibition in neurons leads to a transcriptome remodeling pattern like the one seen in Alzheimer’s disease, and that RBFOX1 overexpression in deficient cells shifts the transcriptome profile back toward its normal state.
In their studies on mRNA stability, investigators in Dr. Najafabadi’s lab have used bulk mRNA, which provides an average measurement of the cellular events but does not capture mRNA degradation differences at the single-cell level, which could be informative about the heterogeneity of mRNA stability across cell types.
“We hope that as single-cell RNA-seq technology moves forward, we can have enough coverage from single cells to look at both mRNA and pre-mRNA to study the rate of RNA degradation at the single-cell level,” states Dr. Najafabadi. “We are currently working on that.”
1. Macosko EZ, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161: 1202–1214 (2015).
2. Habib N, et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nature Methods 14: 955–958 (2017).
3. Wu H, Gordon JAR, Whitfield TW, et al. Chromatin Dynamics Regulate Mesenchymal Stem Cell Lineage Specification and Differentiation to Osteogenesis. Biochimica et biophysica acta. 2017;1860(4):438-449. doi:10.1016/j.bbagrm.2017.01.003.
4. Alkallas R, Fish L, Goodarzi H, Najafabadi HS. Inference of RNA decay rate from transcriptional profiling highlights the regulatory programs of Alzheimer’s disease. Nature Communications. 2017;8:909. doi:10.1038/s41467-017-00867-z.





