
December 1, 2011 (Vol. 31, No. 21)
Richard A. A. Stein M.D., Ph.D.
Combination of Experimental and Computational Views Brings Tremendous Power
The 1953 discovery of the DNA double helix, the 1995 sequencing of the first genome of a free-living organism, and the 2001 publication of the first human genome draft, catalyzed by technological and methodological developments, are among the major advances that helped unveil a remarkable amount of information about many organisms. While these developments helped characterize individual components of the cells in unprecedented detail, they often failed to capture the complex interactions that these components establish as parts of intricate biological networks.
Systems biology, a multidisciplinary approach essential for understanding how these individual parts are organized and function in live cells, has emerged as a powerful field that provides a higher level analysis and promises new perspectives in the biomedical arena.
“The golden target of systems biology is to construct, in silico, a model of the living cells,” said Eytan Ruppin, M.D., Ph.D., professor of medicine and computer science at Tel Aviv University. Dr. Ruppin’s lab focuses on genome-scale metabolic modeling, a field that initially emerged in bacteria and subsequently expanded to eukaryotic organisms, including humans and, most recently, plants. “These models enable us to use molecular-omics data to predict the metabolic state of an organism with fairly reasonable success and accuracy,” explained Dr. Ruppin.
While genome-scale metabolic modeling allows large numbers of experiments to be conducted within a short time frame and at very low costs, there are still many challenges ahead. “We don’t know the kinetic parameters of most metabolic reactions, even in E. coli,” said Dr. Ruppin.
“But we are making some approximating assumptions, and mathematical modeling frameworks enable us to build metabolic models to describe what is going on inside living cells. Metabolism is the only field today in computational systems biology where we have come close to reasonable genome-scale models, and this approach represents a very powerful tool,” he added.
Dr. Ruppin’s lab made seminal contributions toward advancing our ability to build and utilize genome-scale models of human metabolism. A recent effort that Dr. Ruppin and colleagues focused on is cancer cell metabolism. The concept that metabolic pathways are altered in malignant tumors has been known for decades.
In the 1920s, Otto Warburg, who was awarded the 1931 Nobel Prize in Physiology or Medicine for his work on cellular respiration, unveiled that most cancer cells have a high rate of glycolysis and lactate production under nonoxidative conditions, as opposed to normal cells that generate energy mainly by the oxidative breakdown of pyruvate.
“This concept has been lying dormant for many years, and only in the last decade did a renewed interest emerge toward studying the altered metabolism of cancer cells and exploit it to develop selective therapeutics,” explained Dr. Ruppin.
Dr. Ruppin and colleagues recently developed the first generic model of cancer metabolism, and in collaboration with Eyal Gottlieb’s lab from the University of Glasgow and Tomer Shlomi’s group from the Technion, identified, computationally and experimentally, a compound that selectively renders a specific type of renal cancer cells nonviable.
“Predictions generated through the metabolic model that we built were subsequently validated experimentally, providing a very nice proof of concept that one can use this approach to predict selective drug targets that can be tested, first in cell lines and later in animal models and clinical studies,” Dr. Ruppin said.
An even more recent development that Dr. Ruppin presented at Gordon Research’s “Cellular Systems Biology” conference recently held in Davidson, NC, is a new algorithm to predict metabolic targets that can reverse the metabolic state of aging tissue. One of the strategies to reverse the metabolic state, caloric restriction, was shown to work in several organisms. While the magnitude of its impact diminishes as one ascends through the phylogenetic tree, a considerable effect is observed even in mammals.
Animal models revealed that not only does caloric restriction extend the lifespan, but the muscle gene-expression pattern in older mice becomes similar to the one observed in younger animals. “We used a computational approach to predict genetic perturbations that most efficiently reverse the metabolic state from the old state to the young state, and we are currently working on experimentally validating these predictions,” explained Dr. Ruppin.
mRNA
“We are interested in RNA metabolism, specifically in mRNA splicing, but this should not be studied in isolation from other cellular processes,” said Jean Beggs, Ph.D., Royal Society Darwin Trust Professor at the Wellcome Trust Centre for Cell Biology, University of Edinburgh.
At the meeting, Dr. Beggs described an experimental approach that her lab developed to better understand the kinetics of splicing. While the proteins and protein complexes involved in splicing are relatively well characterized, the dynamics of the biological reactions involved, and their interplay with other cellular events, have been less thoroughly explored, due to several limitations, one being the lack of good quantitative assays for RNA and another being a lack of in vivo kinetic data.
“We don’t know the cellular volume that is available for mRNA, and therefore, we cannot measure mRNA concentrations, but we can estimate mRNA abundance in the cell, and this is the approach that we have taken,” explained Dr. Beggs.
By using the budding yeast as a model, investigators in Dr. Beggs’ lab developed a high-resolution biochemical assay in which quantitative real-time PCR was used to measure the cellular abundance of mRNA as an average number of copies per cell, and the data was validated with single-molecule fluorescence in situ hybridization (in collaboration with Edouard Bertrand, CNRS), which is informative of transcript copy numbers in individual cells. “This approach generated high-resolution kinetic data, better than what was obtained previously,” said Dr. Beggs.
This approach revealed that splicing occurs co-transcriptionally, before transcription is completed, and mathematical modeling showed this is more efficient than post-transcriptional splicing.
By using chromatin immunoprecipitation to monitor RNA polymerase chromosomal localization, Dr. Beggs and colleagues found repeated pauses, lasting approximately 30 seconds, which occur after inducing transcription of an intron-containing reporter gene. These transcriptional pauses coincide with the recruitment of splicing factors, and indicate the existence of a transcriptional checkpoint for splicing at the 3´ end of introns.
“There has been evidence from eukaryotes, indicating that chromatin possibly affects splicing and, more recently, there have been tentative indications that splicing may cause chromatin modifications, and all these indicate that chromatin, transcription, and splicing are potentially coupled,” noted Dr. Beggs.
Traditionally, proteins have been depicted as folding into unique structures allowing them to interact with other macromolecules and perform their activities. However, advances from recent years challenge this view.
“Our group focuses on proteins and protein regions that do not fold into a unique structure, and these are called disordered or intrinsically unstructured proteins,” said Péter Tompa, M.D., Ph.D., professor and director of the department of structural biology at Vrije Universiteit Brussels.
Researchers in Dr. Tompa’s lab and other groups unveiled many intrinsically unstructured proteins; elucidating their involvement in the interactome from a systems biology perspective becomes a crucial task.
“Our lab and several other groups have shown that structural disorder is very much involved in the interactome, in the sense that proteins organizing the interactome, and having many other binding partners, are often more disordered than other proteins,” said Dr. Tompa.
An interesting feature of proteins that can adapt to different binding partners is that their function is not as unequivocal as it is for folded proteins. While the function(s) of folded proteins can be clearly defined, disordered proteins bring an element of promiscuity or ambiguity, as they often bind different partners and may perform different functions with each of those partners. “This is what we call moonlighting,” explained Dr. Tompa.
In recent years, Dr. Tompa and colleagues revealed that, as part of this moonlighting activity, certain intrinsically unstructured proteins may have both inhibiting and activating effects on different binding partners or even on the same binding partner—for example, the Epstein-Barr virus SM protein can function both as a post-transcriptional inhibitor and as an activator of gene expression.
Moonlighting increases the functional complexity of the proteome because one protein may have different locations and perform several different functions within the cellular protein network. This concept challenges the classical notion in molecular biology that a gene encodes a protein with a certain structure, which in turn serves a specific function.
“Exceptions exist at many points: one polypeptide sequence may not encode for a structure but for an ensemble of structures, and this ensemble of structures may perform many functions and govern many interactions in the interactome. So the whole view is more complex and more complicated than previously thought,” said Dr. Tompa. “All these aspects have to be incorporated into any systems biology model that is used to describe the cell as a whole,” added Dr. Tompa.
Unveiling the different functions of proteins involved in moonlighting can be challenging, particularly when, after one function is found, it is assumed that it represents the only role of that protein, and other functions might not even be pursued. Cytoskeletal proteins, many of which perform, in addition to their originally described structural roles, enzymatic activities, provide a fascinating example.
Yeast Screens
Jim E. Wilhelm, Ph.D., assistant professor in cell and developmental biology at the University of California, San Diego, and colleagues recently conducted an extensive screen of the yeast GFP strain collection to identify proteins that assemble into intracellular structures that were not captured in previous screens.
“We knew that there are things that the original screens missed,” said Dr. Wilhelm. This more extensive screen unveiled six novel distinct filaments in the budding yeast. “We were particularly excited to find cytidine triphosphate synthase assemble into filament systems.”
Investigators in Dr. Wilhelm’s lab found that an inhibited form of CTP synthase is necessary for assembly into filaments, indicating that enzymatic regulatory activity is linked to incorporation into supramolecular complexes, establishing a link between enzymatic regulation and structural organization of cytoskeletal filaments. Dr. Wilhelm and colleagues subsequently revealed that CTP synthase filaments also exist in the fruit fly, but only in certain tissues, pointing toward the existence of tissue-specific regulation.
“We think that what is going on is very analogous to the principles behind tubulin organization,” Dr. Wilhelm added. Many studies revealed that soluble tubulin subunits do not hydrolyze GTP unless they become incorporated into filamentous structures, in which the plus end of the filament acts as a GTPase activating protein that regulates the enzymatic activity.
Investigators from Dr. Wilhelm’s lab are applying the same principles to study entire metabolic pathways. “This type of regulation is not a random feature but, instead, it seems to be coordinated, and there is a systems biology principle at work,” explained Dr. Wilhelm.
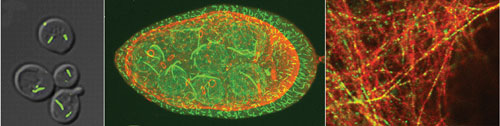
Researchers at UC San Diego conducted an extensive screen of the yeast GFP strain collection. The screen revealed six novel distinct filaments in the budding yeast.
Pathogens
At the meeting, Jason A. Papin, Ph.D., assistant professor of biomedical engineering at the University of Virginia School of Medicine, talked about systems biology approaches that his lab uses.
Another pathogen that Dr. Papin and colleagues focus on is Clostridium difficile. “Particularly in our work on Clostridium difficile, we very much focused on the host side of this interaction, to understand how the intestinal epithelium responds to toxins made by the pathogen,” revealed Dr. Papin.
While many pathogens were characterized at the molecular level, and host factors involved in the interaction with pathogens have also started to be unveiled, albeit at a somewhat slower pace, systems biology now enables investigators to integrate all the information into computational predictive models.
“Characterizing the dynamics of the host-pathogen relationship is an exciting field, and represents one of the opportunities in which systems biology can have one of its first major impacts,” said Dr. Papin.
Systems biology can be used to understand how pathogens behave in a variety of environments or in different mutational backgrounds, or to predict therapeutic targets.
“Within infectious diseases, there are many clinically relevant aspects, such as the challenges that surround emerging pathogens and antibiotic resistance, for which systems biology approaches are becoming increasingly important,” Dr. Papin emphasized.
“The field is moving past metabolism, into the bottom-up network reconstruction of protein synthesis pathways, regulons, and stimulons, and these are important because antibiotic targets and virulence factors can be found in these processes,” said Bernhard Ø. Palsson, Ph.D., professor of bioengineering and adjunct professor of medicine at the University of California, San Diego.
Dr. Palsson and colleagues illustrated the strengths of this approach with the reconstruction of the leucine-responsive E. coli regulon involved in regulating nitrogen metabolism. The analysis revealed multiple distinct regulatory modes for open reading frames and different types of regulatory network motifs, illustrating an approach that can be applied to other organisms to allow the reconstruction of transcriptional regulatory networks.
An important advance in Dr. Palsson’s lab is the recent characterization of the genome-scale metabolic network in several bacteria of biomedical and industrial importance, including Yersinia pestis, Salmonella, and Klebsiella pneumoniae, and the publication of an updated version of a previously released E. coli metabolic network that includes many newly characterized genes and biological reactions and emerges as the most comprehensive metabolic reconstruction of a microorganism to date.
“Looking at transcriptional regulatory networks at the genome scale will become particularly important for infectious diseases because pathogens are constantly sensing the microenvironment, as they decide how to interact with a host,” said Dr. Palsson.
A rapidly expanding field that thrives on a combined experimental and computational perspective, systems biology explores biological processes at levels that were previously impossible to capture. This vibrant discipline marks a paradigm shift from the reductionist approach, which focuses on individual cellular components, to an integrated approach that aims to capture the complex networks established by the interacting components. One of the most relevant portrayals of the transition that systems biology catalyzes, conveying its most important characteristic, is offered by Denis Noble, in his book “The Music of Life”: “[i]t is about putting together rather than taking apart”.

Metabolic network model with nodes accounting for genes, metabolites, and reactions for which the model has a mathematical representation.[University of Virginia School of Medicine]





