
June 1, 2014 (Vol. 34, No. 11)
Kate Marusina Ph.D.
Discovery metabolomics involves the comparison of the metabolome between control and test conditions to find differences in the metabolite profiles. The discovery process comprises three main steps: profiling, identification, and analysis.
Profiling consists of the untargeted acquisition of data under conditions of high analytical reproducibility. Identification involves the use of existing reference standards to annotate detected metabolites. Analysis, which is carried out with statistical software, is matter of sifting through the resulting data.
While this three-step process may seem clear enough, discovery metabolomics is far from straightforward. Indeed, it presents the scientific community with a number of complications. For example, scientists need to find ways of standardizing separation techniques, determining the biological relevance of discovered fragments, and handling the sheer volume of potential metabolites. If discovery metabolomics is to achieve its full potential, scientists will need to stay current, sharing and incorporating the latest technological innovations, tasks that may be accomplished more easily with the benefit of social networking.
Another way to stay current is to read articles such as this one. It explores some of the more interesting applications now emerging in metabolomics. If these applications are any indication, metabolomics is poised to dramatically expand its impact on research and development.
Discovery (nontargeted) metabolomics faces a significant challenge—identifying unknown metabolites in complex biological mixtures. The most common approach consists of searching available databases using exact mass and MS/MS spectral data. “Unlike protein fragments that can be easily differentiated by mass/charge ratio and MS/MS data, metabolite structures are often too similar to each other to be identified on the basis of typical mass spec data alone,” says David F. Grant, Ph.D., associate professor of pharmaceutical sciences at the University of Connecticut.
The identification of unknown metabolites is further complicated by not having all possible metabolites in well-annotated reference databases such as the Human Metabolome Database (HMDB). Another database, PubChem, contains millions of candidate chemical structures, but only a small proportion of them have biochemical significance.
Dr. Grant’s lab is working on three complementary approaches to allow identification of endogenous mammalian metabolites during the initial discovery phase. The first approach expands existing metabolite databases by including structures derived in silico. The team used the assumption that common Phase I drug metabolizing enzymes, such as cytochrome P450, metabolize endogenous compounds. Using well-known biotransformation rules, they computationally generated over 400,000 anticipated metabolites not previously found in any other existing database.
Dr. Grant’s second approach is to add additional filters to narrow down the possible candidate metabolites identified from their In Vivo/In Silico Metabolites Database (IIMDB) and PubChem. Possible matches are evaluated with MolFind software, which relies on HPLC/MS data. Besides mass/charge ratios and MS/MS spectral data, HPLC/MS parameters include retention time, drift time, and ECOM50. The initial set of structures is sequentially sorted by comparing experimentally derived features with computationally predicted features.
The last approach, which is followed after the last filter, involves passing the remaining candidate compound set through a bioinformatics tool called BioSM. This software was “trained” on known mammalian endogenous biochemical structures to identify biochemical structures in chemical structure space. BioSM predicts whether a given structure is consistent with biochemical scaffolds, and therefore could represent a real human metabolite.
“Our software tools lay an important foundation for future metabolite discovery,” continues Dr. Grant. “We hope the research community will continue to standardize discovery technologies and protocols to take full advantage of our computational models.”
Metabolon develops commercial applications of metabolomics technologies. Its comprehensive platform covers the continuum from discovery to diagnostics.
“Metabolon’s discovery approach solves many of the key challenges in metabolomics analysis. Our platform automatically identifies known and novel metabolites in a biological sample while simultaneously ensuring the noise is effectively removed,” says Steve Watkins, Ph.D., the company’s chief technology officer. “One of the key innovations is proprietary software. It accounts for all ion features in a biological sample and compares these features to an authenticated chemical library of thousands of biochemicals.”
After global profiling reveals involvement of certain biochemical pathways, Metabolon offers focused quantitative assays for each intermediate in the pathway of interest. The comparative metabolomics study of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis (NASH) provided multiple insights into lipid metabolism presented in these diseases. While the two diseases are physiologically similar, their global metabolic profiles revealed key differences.
In particular, peroxisomal dysfunction was found only in the pathogenesis of NASH. Because biochemical pathways are well understood, the analysis of metabolic perturbations may serve as a basis for hypothesis-driven studies of the specific biochemical nodes.
To do that, Metabolon offers precise targeted assays for the critical compounds. “Targeted assays are always developed under custom specification,” continues Dr. Watkins. “We also pursue development of our own CLIA diagnostic assays.” Two of Metabolon’s diagnostic assays are currently marketed via its clinical lab partners. Quantose™ provides a quantitative measure of glucose metabolism as a predictive risk assessment tool for those at risk of developing type 2 diabetes.
Dr. Watkins points out that one of the most exciting opportunities for metabolomics is to provide insights into molecular mechanisms of dysregulations defined by a “static” biomarker, such as a nucleotide polymorphism.
Genome-wide association studies (GWASs) identify disease-specific genomic loci, but this is not sufficient to understand the function of the underlying biological processes. Linking genotypes to metabolic signature can shed light on how a genome works. Associations of genomic loci with metabolic traits could be used to gain novel information about possible metabolic changes associated with biological processes underlying that association.
To support new discoveries in this space, Metabolon began collaborating with Human Longevity. Metabolon’s metabolic profiles will supplement whole-genome sequencing and help to make inroads in research on aging and age-related diseases.
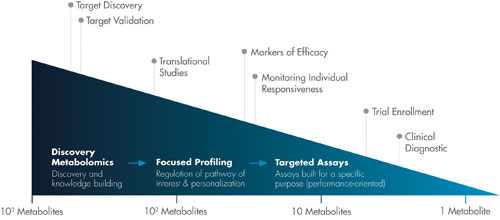
Metabolon’s platform for metabolomic analysis, which covers the continuum from discovery to diagnostics, offers focused quantitative assays for each intermediate in the pathway of interest.
Global Metabolome Changes
“Differentiation of the metabolites of biological origin from artifactual signals remains a challenge,” contends Chris Beecher, Ph.D., chief science officer at IROA Technologies (formerly NextGen Metabolomics) “A large portion of the observable signal in mass spectrometry is simply chemical ‘junk’ unrelated to biochemical processes.” The company continues to perfect IROA™, an MS technique that filters out chemical “noise,” leaving only biologically relevant molecules.
IROA technology has its roots in a physiochemical phenomenon of naturally occurring C13 atoms. C13 has an extra neutron and produces a pattern of additional peaks for each metabolite in a sample. Because of the low natural abundance of C13, these additional peaks (called M+1, M+2, etc.) are small and not very informative.
“We boost the C13 concentration by growing cells in a culture media engineered with precise balances of C13-bearing components,” explains Dr. Beecher. “When comparing an experimental culture grown in 5% C13 with a control culture grown in 95% C13, each metabolite will be represented by a mirror pattern of IROA peaks. This mirror pattern equivocally traces molecules of biological origin to each cell culture, and separates them from nonbiological artifacts that do not have mirror patterns.”
“Because of these unique patterns, control and experimental samples can be pooled and analyzed simultaneously, and their metabolite levels directly compared with a significant reduction in error,” continues Dr. Beecher. “IROA peaks also provide information about mass and the number of carbons. Together, these parameters enable exact prediction of the formula.”
As an example, the company used the IROA symmetry for the global metabolic analysis of Caenorhabditis elegans. The worms were fed bacteria grown on 5% C13 culture or 95% C13 culture. Isotopes incorporated into every molecule of the worm. Such isotopically labeled worm lysates demonstrate recognizable IROA mirror patterns for all biological compounds contained in the organism.
Next, the worms were subjected to heat shock or oxidative stress. Automated measurements of relative concentrations of metabolites under treatment conditions revealed considerable disturbances in the C. elegans endometabolome, including the purine synthesis pathway.
“This experiment demonstrates the ability of IROA to serve experimental needs at the level of higher organisms,” asserts Dr. Beecher. “We continue to move this technique up the evolutionary ladder to mammalian cells, and then, potentially, into clinical research.” According to Dr. Beecher, the ability to identify biologically relevant metabolites, along with their mass and the number of carbons, is a fundamental tool for any discovery platform.
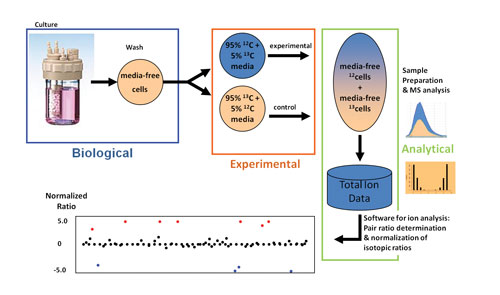
In a method developed by IROA Technologies, isotopically labeled cells are grown and then treated experimentally. When pooled cells are processed, the signals for the compounds from both the control and experimental cells may be distinguished. Artifacts are discarded, and the respective areas for control-derived and experimentally derived compounds are identified (by isotopic signature) and compared (by ratio). The distribution of ratios is analyzed for outliers. Compounds with abnormal ratios are molecules affected by the experimental stressor, facilitating interpretation.
Secrets of Microbial Colonies
The existing metabolomic databases are insufficient to cover the unique molecules produced by even the most common microbes. “We need alternative methods to look at data fragmentation for improved insights into spatial and temporal relationships between biological systems,” says Pieter Dorrestein, Ph.D., director of the Mass Spectrometry Therapeutic Discovery Center, University of California, San Diego.
Dr. Dorrestein’s team uses spectral alignment algorithms (co-developed with the UCSD Center for Computational Mass Spectrometry) that enable visualization of all the fragmented molecules in one layout. The team also uses Cytoscape, a sophisticated software developed by the systems biology community. This software clusters fragmented molecules and arranges the clusters into molecular networks. Once the spectra are organized based on fragmentation similarity, one can identify analogs and related compounds.
The team provides the network tools and the data repository as web resources and makes all their data available publically. “We invite the scientific community to contribute to the collective annotation of molecular networks,” indicates Dr. Dorrestein. “In other words, we are crowd sourcing molecular characterization. Such a community effort is required to ensure the future of metabolite research.”
A comparison of microorganisms with mutated gene clusters could zoom in on metabolites encoded by the gene cluster of interest. “Many useful microbial compounds are produced via nonribosomal synthesis,” continues Dr. Dorrestein. “Often, these are transient and, therefore, difficult to detect. We developed an effective approach that allows metabolic profiling of live colonies directly from a petri dish without any intermediate sample-preparation steps.”
Nanospray desorption electrospray ionization (nanoDESI) setup delivers solvent precisely to the spot of interest, such as an immediate area around a bacterial colony. The capillary of the nanoDESI is connected with the MS nanospray capillary by a liquid droplet, which forms a bridge between the two capillaries. The analyte is desorbed at the liquid bridge and immediately transferred into electrospray of the mass spectrometer.
Dr. Dorrestein points that this methodology allows investigators to home in on molecules of biological relevance. Moreover, this elegant approach significantly reduces the man-years needed for identification of new metabolites.
Dr. Dorrestein plans to expand this methodology to understand relationships between microbes and human tissues such as skin, lungs, or gut. His team began with a topographical analysis of skin. Microbial and molecular data resulted in a molecular geography rich with information about relationships between humans and their microbiota.
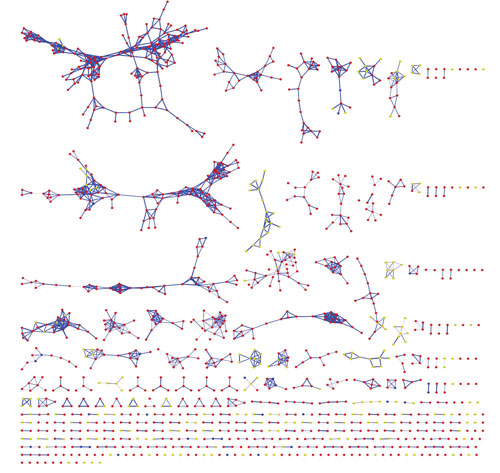
Researchers at the University of California, San Diego are extending their methodology, which enables the visualization of fragmented molecules in one layout, to understand relationships between microbes and human tissues. In this image, data generated by Neha Garg, Ph.D., is used to compare metabolites from lung and coral microbial communities.





