
June 15, 2012 (Vol. 32, No. 12)
Neil McKenna, Ph.D.
Small molecule inhibitors (SMIs) are well-established therapeutics in the pharmaceutical industry. Some, such as Tamoxifen, an inhibitor of estrogen receptor function that is widely used in breast cancer, and flutamide, a prostate cancer therapeutic that inhibits androgen receptor signaling, are among the most successful drugs on the market. While previous SMI drug discovery approaches relied primarily on trial and error testing of chemicals, this has been supplanted by recent advances in genomics and proteomics, combinatorial chemistry, and high-throughput screening that have rapidly accelerated the rate at which compounds are entering the drug pipeline. SMIs can be designed to disrupt key eukaryotic cellular processes, such as kinase signaling pathways and cell division, that have applications in cancer and inflammatory disorders. They can also be developed to specifically interfere with viral and prokaryotic biology in the treatment of infectious diseases.
Cambridge Healthtech’s recent “Drug Discovery Chemistry” conference in San Diego brought together researchers from both industry and academia to highlight recent progress in the field. A recurring theme in these talks was targeting protein-protein interactions in small molecule drug discovery.
Protein-protein interactions are fundamental to myriad cellular processes and, by extension, are essential to the progression of many diseases. Interactions between proteins can be inferred from genomics screens for functional relationships or directly from large-scale proteomics screens.
“In many cases, the biology can tell us what protein-protein interactions might be key targets for therapy,” said Michelle Arkin, associate adjunct professor of pharmaceutical chemistry at University of California, San Francisco and associate director of UCSF’s Small Molecule Discovery Center.
“Some of the best targets are extracellular hormone/receptor interactions that initiate intracellular signaling events, and enzymes that act as functional units within protein complex networks,” she added.
Histone deacetylases (HDACs), for example, have emerged as some of the most popular therapeutic targets in recent years. HDACs catalyze the removal of acetyl groups from nucleosomal histones, to convert chromatin to a “closed” state, resulting in the repression, or silencing, of genes. Targeting the interactions between complexes containing these molecules and specific transcription factors can therefore alter the transcriptional function of a target disease cell and compromise its viability.
Karus Therapeutics is developing a series of selective inhibitors of HDAC6 that were the focus of a talk by its CSO Stephen Shuttleworth.
HDAC inhibitors have shown promise in the treatment of rheumatoid arthritis and other inflammatory disorders. Inhibition of HDAC6, for example, leads to increased expression of the transcription factor forkhead box P3, an important regulator of T-cell function in the immune system, and whose deficiency has been linked to autoimmune disorders such as systemic lupus erythematosus.
“Discovery and development of HDAC6 inhibitors has historically been extremely challenging, and there is currently a dearth of their isoform-specific inhibitors,” said Shuttleworth. HDAC6 has also been implicated in the etiology of neurodegenerative disorders such as Alzheimer disease.
Carl Baldick, a senior investigator in infectious disease research and development at Bristol-Myers Squibb (BMS), turned the focus to the inhibition of the hepatitis C virus (HCV). “There are several key challenges in achieving high cure rates in HCV therapeutics,” reported his colleague Douglas Manion, svp of development, neuroscience, virology at BMS.
“These include avoiding the use of interferon-alpha, shortening the duration of therapy and improved safety and tolerability,” Manion added. An additional obstacle is the diversity of viral genotype as well as geography and treatment experience of patient populations. Assembly of the virion involves interaction of the capsid protein with the NS5A regulatory protein. BMS’ daclatasvir, currently in Phase III, inhibits the function of NS5A and has potent antiviral activity across multiple HCV genotypes.
Other HCV therapeutics in BMS’ portfolio include inhibitors of the NS5B polymerase (INX-189 and BMS-791325) and the viral NS3 protease inhibitor asunaprevir. “We are continuing to explore other ways of attacking HCV, including the potential for entry inhibitors that could be combined with and complement other direct-acting antivirals targeting HCV replication,” said Baldick.
BMS has developed a preclinical assay that allows the evaluation of the potency of entry inhibitors against all HCV genotypes. “The assay uses a panel of HCV pseudoparticles containing functional viral envelope proteins derived from 40 different samples from patients infected with HCV genotypes, 1–5,” explained Baldick.
According to Manion, future prospects for HCV SMI development at BMS include an external collaboration with Janssen Pharmaceutica to evaluate combination therapy of aclatasvir and BMS-986094 with the TMC-435 protease inhibitor, and the development of triazine entry inhibitors. “Current triazine entry inhibitors lack sufficient potency against genotypes 2–5, although it is possible that further chemical modifications to this chemotype could meet this goal. In that case, HCV entry inhibitors could be a promising component of combination therapy,” Baldick noted.
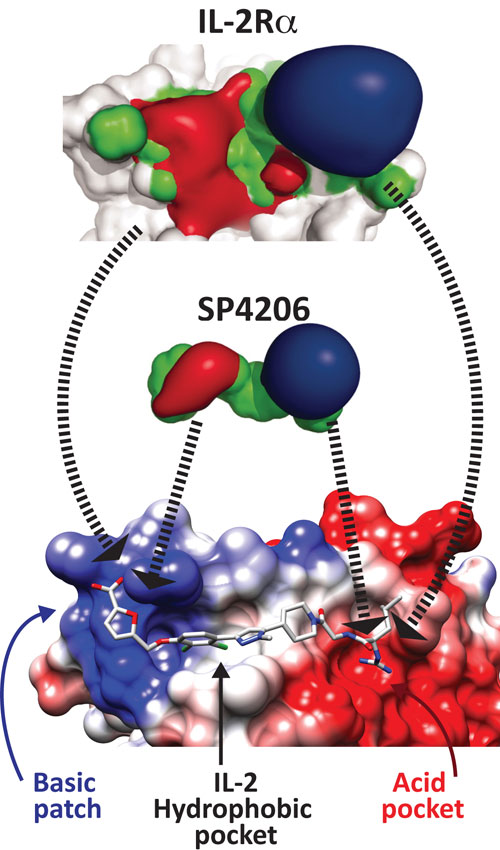
A small molecule inhibitor of IL-2 mimics the IL-2 receptor: Top: binding surface of the alpha chain of the IL-2 receptor, showing negatively charged surfaces (red), neutral (green), and positive (blue). The interface is composed of four distinct regions of secondary structure, and is therefore a highly discontinuous epitope. Middle: the charge distribution on SP4206, a 60 nM inhibitor of IL-2. Coloring is the same as for the IL-2 receptor surface, and shows a striking similarity in surface charge distributions of the IL-2a receptor. Bottom: crystal structure of SP4206 bound to IL-2 at the protein-protein interface. The colors are the same as in the top image; there is charge complementarity between SP4206 or IL-2 receptor and the surface of IL-2. The modular nature of the binding site is also shown. [UCSF/ Adapted from Thanos, et al, PNAS]
Protein-Protein Interactions
Specificity is a challenge with inhibitors of protein-protein interactions—for example, different isoforms or splice variants of a protein can have distinct functions. Arkin stressed the importance of SMIs targeting protein-protein interactions on an individual basis, and such an approach requires a more nuanced approach to SMI discovery.
“We like to take a fragment-based approach at UCSF, where SMIs are discovered in pieces,” Arkin explained. “The potential chemical diversity is much smaller for a fragment, 250 Da, than for a typical drug, 500 Da, thereby reducing the bias inherent in selecting compounds,” she added. Another advantage is that fragment screening encompasses biophysical and structural methods, resulting in higher topographical resolution at the drug-protein interface.
A recently published example of the successful development of high-specificity (nanomolar) inhibitors of Menin-MLL protein-protein interactions was the subject of a talk by the study’s lead author, Jolanta Grembecka (University of Michigan, Ann Arbor). The MLL gene encodes a transcriptional co-activator with important roles in hematopoiesis, and chromosomal translocations of this gene generate chimeric proteins that initiate aberrant programs of gene expression that result in the development of acute leukemia.
The interaction of MLL fusion proteins with menin, a component of a histone methyltransferase complex, is an important step in the development of leukemia and as such is an attractive target for the development of therapeutics in this disease.
Starting with a detailed study of the MLL-menin interaction interface, Grembecka’s study used high-throughput fluorescence polarization screening to identify lead compounds from a library of nearly 50,000 compounds, and identified several candidates that induced cell death in a bone marrow cell model of leukemia.
“There is a lot of activity in the area of protein-protein interaction inhibitors in cancer and immunology because there are many, well-characterized signal-transduction events to inhibit,” observed Arkin. Other well-developed therapeutic targets in this area include programs targeting BclXL at Abbott Laboratories and Genentech and MDM2/p53 at Roche.
Compound Libraries
The SMI drug pipeline relies heavily on the availability of diverse compound libraries that will maximize the number of potential hits in preclinical screens. Roman Kombarov, project manager at Asinex, described ongoing efforts aimed at improving the rate of discovery of novel antibacterial therapeutics.
“The screening of randomly assembled, diverse compound libraries has produced extremely low hit rates,” he pointed out. “Libraries from commercial sources are overpopulated with compounds dedicated to screening against nonbacterial compounds such as GPCRs and nuclear receptors,” he added. The solution, Kombarov suggested, is the development of novel libraries specifically tailored to the discovery of compounds that can kill bacterial cells through effective penetration of their membranes and inhibiting bacterial-specific targets without affecting the viability of mammalian cells.
“Other important factors must also be considered, such as the amenability to follow up with the hits and their chemical novelty and patentability,” he added.
A large proportion of known antibacterials have come from natural products, but the inherent complexity of natural product chemistry has often prevented their progress down the drug pipeline.
“At Asinex we have developed an inverse medchem approach that takes advantage of synthetic-friendly, natural, product-like scaffolds and decorates these scaffolds with carefully selected peripheral residues such as hydrophilic and amphiphilic saturated fused-, spiro-, and macrocyclic systems,” Kombarov reported.
An Asinex compound library developed using this approach recently generated a 2% hit rate when screened against a panel of bacterial pathogens. He said. “We believe our library is capable of delivering multiple excellent starting points for completely new antibacterial chemotypes worthy of further characterization and development.”





