
April 15, 2013 (Vol. 33, No. 8)
Brett Levay-Young
Melanie Olesiuk
Claes Gustafsson Ph.D. COO and Founder ATUM
Jeremy Minshull, Ph.D. DNA2.0
Maximizing Screening Efficiency through Good Design
Proteins with desirable properties are often identified by screening libraries of related gene sequences. Successful applications include antibody engineering, vaccine development, and biocatalyst optimization. Properties as diverse as thermal stability, protein-ligand affinity, kinetics, substrate specificity, and even catalytic mechanism can all be modified using libraries and functional screening.
Libraries are also useful for studying protein sequence/structure-function relationships in biological systems. Libraries may be as small as a handful of variants or as large as 1012 variants or more.
This tutorial provides a brief introduction to guide the choice of library format based on the target application and screening capabilities available for the project. DNA2.0 offers a wide variety of protein variant libraries to meet different research needs—only a sample of library options are described in this tutorial.
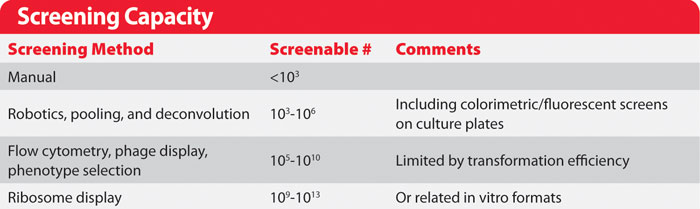
Screening Capacity
Two critical screening factors affect library choice: how many measurements can be made, and how well those measurements reflect the properties that are actually being sought.
As the number of variants screened approaches the theoretical size of a library, the greater the probability that any newly sampled individual has already been sampled. It can be calculated that one must sample three times the total library size to have a 95% chance of testing any particular member of a library. One must further sample 20 times the total library size to have a 95% chance of testing every member of the library.
In practice more variants would have to be screened to completely sample a real library, because libraries can not be made with perfect distributions of variants. This law of diminishing returns means that researchers generally avoid exhaustive library testing. Instead, iterative cycles of library construction and screening enable sparse screening of large libraries and precise testing of small libraries to yield highly improved variants.
Screening Capacity
Random Mutation Libraries
Random errors can be easily and inexpensively introduced into a gene sequence by a variety of methods, the most common of which is error-prone PCR. Mutational frequencies can be controlled from ~0.2 to 5 random substitutions per 1 kb DNA, where the number of mutations has a Gaussian distribution across the library. Libraries on the lower end of the mutation spectrum will encode many wild-type sequences. Even though in principle the theoretical size of a random mutagenesis library is very high, cloning and transformation efficiency of the constructs usually limits sizes to 108–1010.
Substitutions identified by random mutagenesis libraries can subsequently be combined and more carefully assayed in a combinatorial library.
Degenerate Codon Libraries
A more thorough sampling of individual mutations can be achieved by incorporating degeneracies into a nucleic acid sequence at specific codons. Changing a codon to, e.g., NNK (N=A/C/G/T, K=G/T), exhaustively samples all possible amino acid substitutions (and one STOP codon) at the chosen positions. Degenerate codon libraries usually have distribution biases caused by the uneven degeneracy of the genetic code.
For example, Tryptophan is encoded by a single codon, while Serine is encoded by three codons in an NNK library. For libraries in which only one or two residues are varied in each clone this may not be a serious concern. However, as the number of residues sampled increases, the bias in the library escalates. A six-codon NNK library will have 109 (412 x 26) possible nucleotide sequences (including STOP). The probability of finding 6xTrp will be equal to finding 6xSTOP and ~750-fold lower than finding 6xSer.
One popular variation on degenerate codon libraries is a set of variants in which all 19 amino acid substitutions are enumerated at every position in a protein in turn. The library is delivered as individually arrayed variants with defined sequences or as a mixed pool of all 20 possibilities for each position. This greatly reduces the screening burden; it also has advantages in patent filing since it is possible to test every amino acid mutation in a protein.
Combinatorial Libraries
Advances in oligonucleotide manufacturing processes and library-assembly protocols now enable the synthesis of libraries where the distribution of codons at any position can be controlled with reasonable accuracy (Figure 1). The more-refined synthesis methods used to create these libraries also allow control of relative ratios of amino acids at different positions, independent of genetic code degeneracy. Peptide loops or amino acid deletions can also be introduced as variables into these libraries.
Combinatorial libraries can be used to explore large, complex sequence spaces, e.g., in mimicking germline antibody diversity. They can also be used to create informative combinations of mutations identified from random mutation and degenerate codon libraries, or possible substitutions identified through bioinformatics means.
By controlling mutation ratios and minimizing synthesis errors, combinatorial libraries can drastically decrease the number of variants needed to be screened, allowing the use of low-throughput assays that are better indicators of the functionality desired.
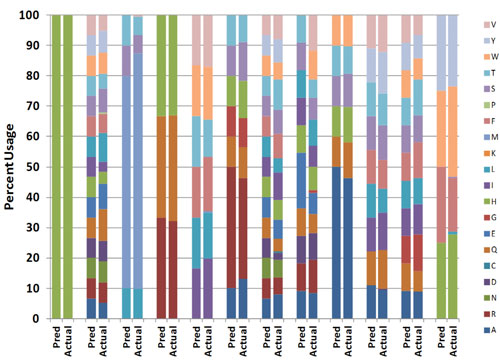
Figure 1. In this combinatorial library, a variable region of an antibody was diversified. Each column pair represents a given residue. “Pred” signifies the desired or predicted diversity. “Actual” represents the codon distribution in the library as determined by deep sequencing. First column pair is the control codon.
Modular Libraries
Modular libraries extend the combinatorial library concept beyond individual amino acid changes to create new genetic constructs from basic DNA parts including regulatory elements and/or coding sequences. Genetic elements are synthesized de novo once and subsequently combined scar-free in predefined combinations or randomly, with the order of elements preserved (Figure 2). These libraries can be used to create new multi-domain proteins, biochemical pathways, genetic circuits, and organisms.
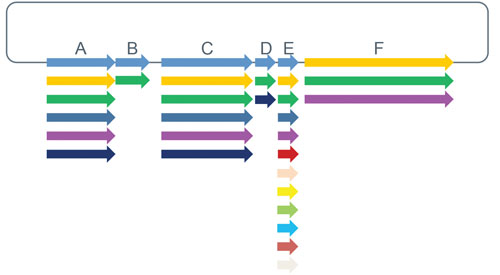
Figure 2. Modular libraries utilize synthesized DNA fragments that are combined using scar-free cloning to explore domain-based biological systems. Modular libraries can be delivered as arrayed, sequenced verified variants, or as a pool of molecules.
ProteinGPS™ “Libraries”
When meaningful measurements of function can only be made in low throughput, optimal sampling of libraries can be calculated. Design of Experiment (DoE) algorithms are used to identify a small set of variants (~100) that are synthesized and assayed to provide the most possible information of the sequence space (Figure 3). The data are then analyzed using multivariate regression methods to tease out sequence-activity relationships. This technique is equally applicable to finding beneficial silent mutations, amino acid changes within a protein, or optimal combinations of higher-order genetic elements in a pathway.
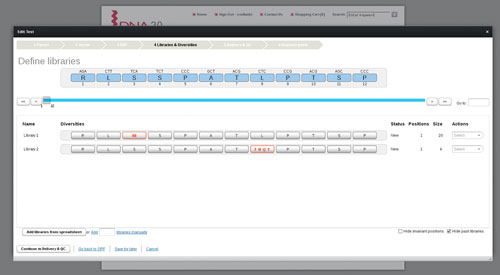
Figure 3. DNA2.0 provides a free online Library Designer tool, allowing researchers to quickly and efficiently design the optimum protein variant libraries for their research goals.
Summary
Exploring and engineering biological systems requires a balance between thorough functional testing and broad sequence exploration. Efforts applied to library design and synthesis can compensate for limitations in screening capabilities, whether these limitations restrict the absolute number of clones that can be tested or the relevance of high-throughput assays to the properties that are required. DNA2.0 provides a portfolio of library formats to meet the needs of biotech projects, ranging from extremely HTP antibody discovery using flow cytometry screening to experimentally challenging, low-throughput, high-quality pathway engineering (Figure 3).
Brett Levay-Young ([email protected]) is protein library senior manager, Melanie Olesiuk is protein library engineer, Claes Gustafsson ([email protected]) is CCO, and Jeremy Minshull is CEO at DNA2.0.

