
May 1, 2012 (Vol. 32, No. 9)
Mikael Kubista, Ph.D. TATAA Biocenter
According to a recent report on the gene-amplification technologies market from Global Industry Analysts, there are close to 70,000 bioresearchers using real-time quantitative PCR (qPCR) in North America alone. They spend $740 million annually on instruments and reagents. Annual growth, notes the market report, is about 16% and may even increase as the major hurdle for small companies trying to enter the field—basic PCR technology patent protection—expired last year. The global market is forecast to reach $1.9 billion by 2015.
Several technological improvements and innovations, as well as new applications and procedures, are driving the field, but there are also roadblocks to overcome for continued growth.
Tissue heterogeneity, where many types of cells respond differently to stimuli, is major complication in biomolecular research. When studying the effect of a certain environmental change or the response to a drug only some of the cells typically respond and they may do so differently. Nonresponsive cells confound the measurement response and obscure analysis.
In other cases the response of a particular rare cell, such as a pluripotent cell or a mutated cell, may be of prime interest. Cells from body fluids can be labeled and sorted on the basis of surface markers using fluorescence activated cell sorting (FACS). Many tissues can be carefully disintegrated into individual cells for FACS analysis.
A sorter can then be used to deposit individual cells of the appropriate kind for further analysis by qPCR. Other means to extract individual cells are aspiration and laser capture.
Once individual cells have been collected, reagents compatible with downstream reverse transcription and qPCR are available for gentle lysis. Also, robust methods for pre-amplification are available. Pre-amplification is a step in single-cell expression profiling that is performed after reverse transcription but before qPCR to amplify the cDNA moderately.
Typically multiplex PCR runs for a limited number of cycles. This produces a sufficient number of copies of each cDNA so that the sample can be aliquoted for parallel singleplex PCR analysis of all the targeted genes. With this workflow up to 96 transcripts per cells have been measured in several studies related to early development and differentiation.
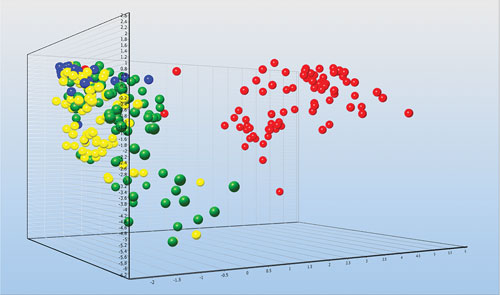
Individual astrocytes from injured mice brains collected by FACS and classified based on the expression of 47 genes: Undamaged brain (blue), three days after injury (yellow), seven days after injury (green), and 14 days after injury (red). The profiling reveals expression pathways activated by the injury that induce the healing process. [Graph made with GenEx. Courtesy of Vendula Rusnakova and Miroslava Anderova.]
Integrated Work Flow
Pre-amplification is, of course, not limited to single cells, but can be used any time the amount of starting material is limited. This makes it possible to study the expression of a reasonably large number of transcripts per sample, and we already see more usage of qPCR in exploratory phases, where the objective is to identify candidate markers for subsequent confirmatory studies.
Of course, qPCR is not close to whole transcriptome profiling, which in the future will be done using microarrays or, even more likely, next-generation sequencing.
Major efforts by leading qPCR companies are focused on developing carefully selected, optimized, and validated assays in sets of 96, 384, and even 2×384 for affordable screening. As opposed to whole transcriptome analysis, this type of selected screening is performed on a larger number of samples than the number of genes analyzed. This makes the analysis more robust.
There are a number of dedicated tools available to identify markers for validation. For example, MultiD Analyses, a company that I co-founded, develops GenEx software for qPCR data mining with multivariate strategies.
With methods such as principal component analysis (PCA), hierarchical clustering, self organizing maps (SOM), and support vector machines (SVM), the optimum set of markers that distinguishes between classes of samples is selected based on the genes’ combined expression profiles. This is a much more powerful approach than selecting markers individually based on differential expression only.
Starting with a smaller number of preselected markers (than essentially the whole transcriptome) has provided the important advantage that false positive rates are greatly reduced and confounding noise is substantially smaller. Important markers not present in the original set can be identified by correlation based on function, property, or disease mining databases. Two of the most powerful products for studying qPCR data are IPA and the iReport, both from Ingenuity Systems.
Standardization and Quality Control
Although companies compete on achieving the highest reproducibility on their qPCR instruments, the qPCR measurement for molecular diagnostics is hardly ever the most confounding step of the testing protocol, provided an adequately performing instrument is used.
Collection, transport, and storage of samples, nucleic acid extraction, and reverse transcription (in case of RNA) are steps that contribute much more confounding variation than qPCR. For example, blood collected in unstabilized matrices, such as EDTA tubes, shows seriously perturbed expression profiles due to the cells’ response to the foreign environment in the test tube. Serious preanalytical problems are also experienced in preserved tissue samples, where common fixation reagents such as formalin, selected primarily to preserve sample morphology, cause serious damage to the RNA.
In Europe a major effort has been undertaken by the SPIDIA consortium to tackle the standardization and improvement of pre-analytical procedures for in vitro diagnostics. The program covers steps from the development of evidence-based guidelines to the identification of quality biomarkers and tools for the pre-analytical phase.
Recently, SPIDIA presented an improved procedure for tissue preservation that maintains RNA at high quality, while preserving morphology. SPIDIA also coordinates proficiency ring trials in Europe helping routine laboratories evaluating their performance in the handling of and analyzing biological specimens with qPCR.
The European Committee for Standardization (CEN) is a SPIDIA member and the consortium interacts closely with the Office of Biorepositories and Biospecimen Research (OBBR) of the National Cancer Institute and the Clinical Laboratory Standards Institute (CLSI).
CLSI has released several guidelines relevant for molecular testing, many of which has been adapted by the FDA. These initiatives are most important to establish qPCR as a robust and reliable platform for routine diagnostics.
For research applications the “MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments” have been developed to encourage better experimental practice and allow for more reliable and unequivocal interpretation of qPCR results.
Several leading journals enforce MIQE and many more endorse them. Guidelines are good but not sufficient; we also need standards. Recently, the U.S. National Institute of Standards and Technology released the first viral DNA Standard Reference Material—for cytomegalovirus (CMV). The CMV was quantified as genome copies/volume using digital PCR (dPCR).
In dPCR a sample is aliquoted into a large number of reaction chambers such that only some contain targeted DNA and give rise to product upon PCR amplification. Counting the number of positive reactions essentially corresponds to counting single DNA molecules, resulting in absolute quantification and traceability to the SI unit, the mole.
qPCR technology is developing rapidly, although most current efforts are on upstream (pre-analytic) and downstream (data mining) processes, rather than on the qPCR technique itself which, I believe, has almost reached perfection.
A lot of work remains to be done until the entire workflow is standardized. Even more challenging will be to develop standards for various important DNA and RNA targets. The guidelines under development now are an important step in the right direction.
Digital PCR Products Continue to Emerge
Joining the firms that have been launching digital PCR technology is RainDance Technologies. The company recently introduced its new RainDrop™ digital PCR system, which enables digital answers across a number of applications including low-frequency tumor allele detection, gene expression, copy number variation, and SNP measurement.
The RainDrop system, built on RainStorm™ picodroplet technology, generates up to 10 million picoliter-sized droplets per lane. Since each droplet encapsulates a single molecule, researchers can determine the absolute number of droplets containing specific target DNA and compare that to the number of droplets with background wild-type DNA. The instrument employs a two-color-per-marker approach and varying probe intensity method that is capable of multiplexing up to 10 markers.
In a recent Lab on a Chip paper, scientists from Université de Strasbourg and Université Paris Descartes used the RainDance digital PCR technology to detect a single mutated copy of KRAS in a background of 200,000 wild-type copies. “With RainDance digital PCR, we were able to achieve absolute quantification of mutated and tumor-circulating DNA and improve the detection of a circulating tumor by comparing its proportion to nontumor DNA,” says Pierre Laurent-Puig, M.D., Ph.D., of the Université Paris Descartes. “Absolute quantification is critical, especially in research that lays the groundwork for future clinical applications, because it allows you to generate meaningful thresholds that will be required for prognostic and diagnostic tools.”
Last fall, Life Technologies launched its digital PCR instrument, the Applied Biosystems QuantStudio™ 12K Flex Real-Time PCR system. This genetic analysis instrument can generate more than 12,000 high-quality TaqMan® data points per run, or up to 110,000 data points in an eight-hour workday, the firm reports. Users can also run digital PCR experiments on the instrument using nanofluidic consumables and dedicated analysis software for increased sensitivity and specificity.
The nanoliter volumes required to conduct experiments on the QuantStudio 12K Flex OpenArray® plates make the instrument cost efficient, saving both reagent and samples, Life Tech notes. The system can accommodate any one of five different interchangeable blocks (OpenArray plates, TaqMan Array Card, 384-, 96 fast-, and 96-well plates) to match the size and type of experiment.
RainDance Technologies’ RainDrop generates up to 10 million picoliter-sized droplets per lane. Each droplet encapsulates a single molecule.
Fluidigm is also a player in the digital qPCR arena. The firm uses its integrated fluidic circuits (digital array chips) to perform digital PCR on up to 48 unique samples per chip, with each sample being partitioned into 770 individual reaction chambers. Fluidigm has experimental chip formats that can change the number of samples per chip, as well as the number of reaction chambers per sample. With the company’s approach, a positive signal indicates the presence of a target, while the absence of signal indicates the lack of a target.
Fluidigm’s digital PCR system is closed, minimizing the possibility of PCR-induced contamination. The firm says the workflow is simple; a single hands-on step is required per sample.
Bio-Rad Laboratories offers the QX100™ Droplet Digital PCR™ (ddPCR) system. The company says that this system provides an absolute measure of target DNA molecules. Applications include enriching for rare target sequences, detecting small fold (1.2x) target differences, and determining copy number without a standard curve.
Bio-Rad reports that the instrument’s sensitivity and precision arise from ddPCR’s ability to partition a sample into 20,000 droplets. Droplet Digital PCR allows researchers to detect 0.001% mutation fractions, completely resolve copy number variations, distinguish up to 10% differences in gene copy, and accurately quantify genes that differ by a single nucleotide, according to the company.

Fluidigm uses its integrated fluidic circuits to perform digital PCR on up to 48 unique samples per chip, with each sample partitioned into 770 individual reaction chambers.
Mikael Kubista, Ph.D. ([email protected]), is a professor, founder, and CEO of the TATAA Biocenter. Dr. Kubista is also a member of GEN’s OMICS advisory board. In addition, he serves as head of the department of gene expression at the institute of Biotechnology of the Czech Academy of Sciences.

