
October 1, 2016 (Vol. 36, No. 17)
Kristine Angevine Product Manager I Rubicon Genomics
Edward Jan Senior Product Manager Rubicon Genomics
Matt Carroll Senior Director Rubicon Genomics
Jinglan Zhang Clinical Molecular and Biochemical Geneticist Baylor Miraca Genetics Laboratories
Hongzheng Dai Baylor Miraca Genetics Laboratories
Richard Yim Doctoral Researcher University College London
Shawn Quinn Partner Curio Genomics
ThruPLEX® Tag-seq Kit Reduces False Positives for Discovery of True Mutations
Research in next-generation sequencing (NGS) is rapidly evolving, especially in several areas of cancer research that rely on detection of mutations. In contrast to germline mutations, somatic mutations may be present at low frequencies within an excess of DNA from normal cells.
Examination of cell-free DNA (cfDNA) is a non-invasive method to study the presence of circulating tumor DNA, which contains somatic mutations. Thus, the ability to detect low-frequency alleles in cfDNA is crucial to many research applications including the tracking of tumor recurrence and characterization of tumor clones.
NGS methods suffer from technical drawbacks that pose challenges for low-frequency mutation detection. Most commonly, base errors are generated and accumulated through the library preparation and sequencing process, making it impossible to accurately reconstruct the sequence of the original molecules and differentiate real mutations from false positives introduced during amplification, sequencing, and base-calling steps. As a result, it is difficult to identify true mutations, because they are hidden by false base calls, also known as false positives.
To overcome these challenges, short, random DNA sequences, named unique molecular tags (UMTs), can be used during library preparation to label input DNA fragments. This method allows for the removal of background errors during data processing so true mutations can be determined. Rubicon Genomics’ ThruPLEX Tag-seq kit is an Illumina® NGS library preparation kit that contains over 16 million UMTs to provide confident mutation calls and has a single-tube, three-step workflow.
ThruPLEX Tag-seq Workflow
In the first step, DNA fragments are end-repaired in a highly efficient process. Following repair, proprietary ThruPLEX stem-loop adapters containing UMTs are ligated to DNA fragments and extended. The UMTs allow individual DNA fragments to be tracked throughout the library preparation and data analysis process. In the final reaction, indexing primers containing Illumina P5 and P7 sequences are used to complete the library structure and amplify the library fragments. Sample indexes—either single or dual—are incorporated to allow pooling and multiplexing of samples during sequencing.
The complete ThruPLEX Tag-seq library structure consists of P5/P7 sequences for flow cell attachment, sample indexes, sequencing primer binding sites, and UMT sequences. Each UMT consists of six degenerate bases, providing 4,096 unique sequence permutations. Together, the UMTs on either side of a fragment produce more than 16 million tag combinations, ensuring that all fragments are labeled uniquely for identification during data analysis. This process allows reads with the same UMT to be grouped computationally into amplification families to determine real mutations, correcting errors generated through library preparation and sequencing.
To build the consensus sequence, mutations present in all reads are called true, while those present in only some reads are false. Consensus sequence reads are then constructed to yield one read per original molecule that is free of library preparation and sequencing errors, correctly reflecting the sequence of the initial molecule. In addition, during data analysis, UMTs allow the original DNA molecules to be counted. DNA fragments sharing the same UMT belong to the same family and can be traced to the original molecule. Therefore, the number of unique DNA molecules can be determined by counting the number of UMTs in the library. After error correction, the resulting consensus sequence is then used to detect low-frequency mutations with high sensitivity and specificity (Figure 1).
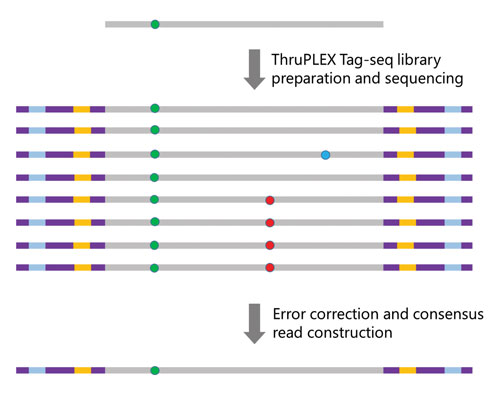
Figure 1. Determining true mutations. The ThruPLEX Tag-seq Kit uses UMTs to label the initial molecule containing the mutation (green dot) at both ends. After library preparation and sequencing, PCR artifacts (blue dot) and sequencing errors (red dot) can be distinguished bioinformatically from the initial molecule, allowing the detection of true mutations and reducing the number of false positives.
Sensitive Variant Detection
To establish the level of variant detection using the ThruPLEX Tag-seq Kit, we measured the limits of variant detection using reference standards engineered with variants present at various allele frequencies. Using 10 ng and 30 ng of cfDNA Horizon reference standards, Baylor Miraca Genetics Laboratories and University College London generated libraries using ThruPLEX Tag-seq, followed by target enrichment to 5,000X coverage and sequencing on a HiSeq® 2500 or NextSeq® 500 system.
Data processing and analysis were conducted using the ultra-fast Curio bioinformatics platform. Background errors (false-positive calls) due to PCR artifacts and sequencing errors were dramatically reduced with ThruPLEX Tag-seq library preparation (Figures 2A, 2B). Consequently, signal-to-noise ratio was also significantly improved (Figure 2C), providing more confident variant detection.
To determine the level of variant detection, six variants at different allele frequencies were examined to compare the expected mean allele frequency (MAF) to the detected allele frequency. Variants were called at their expected frequencies down to 0.5% with 100% sensitivity and >99% specificity (Table 1). Lower detection limits can be achieved, depending on sample quality, input amount, capture efficiency, sequencing depth, and data-processing algorithms.
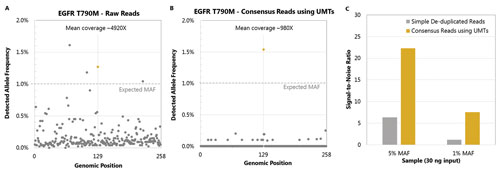
Figure 2. Background reduction using UMT. Data from the same sample was processed without (A) or with UMTs (B). (A) In conventional library prep, true mutations (yellow dot) can be difficult to detect due to background errors (gray dot). (B) With the use of UMTs, the background is significantly reduced and the true mutation can be detected. (C) The signal-to-noise ratio is significantly improved at various allele frequencies between conventional library preparation (gray) and library preparation containing UMTs (yellow).
Conclusion
This data demonstrates that ThruPLEX Tag-seq, equipped with more than 16 million UMT combinations, is a powerful tool for confident detection of low-frequency variants. Variants at 0.5% allele frequency or lower can be detected with high sensitivity and specificity using just 10 ng of input DNA. The combination of ThruPLEX Tag-seq’s highly efficient chemistry and single-tube workflow preserves molecular complexity, allowing researchers to discover more information from precious samples. Researchers have the freedom to use any commercially available capture panels, or to design custom capture panels to interrogate genomic regions of interest that span hundreds of genes and study variants present at low allele frequencies.
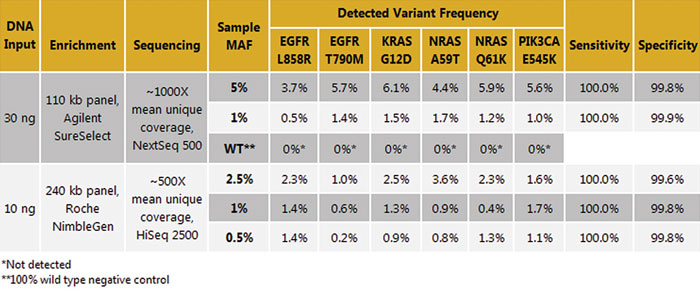
Table 1. Variant frequency detection. Horizon Multiplex I cfDNA Reference Standards (Horizon HD780) were used as is or titrated using the wild-type reference standard to generate samples at additional allele frequencies. Variants were detected at their expected frequencies with high sensitivity and specificity.
Kristine Angevine ([email protected]) is product manager I, Edward Jan is senior product manager, Matt Carroll is senior director of internal operations and bioinformatics at Rubicon Genomics. Jinglan Zhang is clinical molecular and biochemical geneticist and Hongzheng Dai is ABMGG clinical molecular genetics & genomics fellow at Baylor Miraca Genetics Laboratories. Richard Yim is a doctoral researcher at University College London. Shawn Quinn is partner at Curio Genomics.





