
June 1, 2010 (Vol. 30, No. 11)
Michael McGinley senior bioseparations product manager Phenomenex
Greg Scott
Brian Rivera
Simplifying the Preparation and Assessment of ADME/Pharmacokinetic Samples
As candidate oligonucleotide therapeutics move through the drug-development path past compound synthesis and in vitro testing, there has been an increasing need to understand how such compounds behave in vivo. ADME/pharmacokinetics studies require analysis of the chemical structure and quantity of therapeutic oligonucleotides and their metabolites from the time they are introduced into a subject to the point where they are no longer detected in plasma or tissues. These studies are typically performed using liquid chromatography/mass spectrometry (LC/MS).
With small molecule therapeutic analysis, a simple isolation step is usually required to separate the active pharmaceutical ingredient (API) from biological matrices such as plasma or urine. This process is more difficult with oligonucleotides, which are present in small amounts in plasma that is full of proteins, salts, lipids, and cell debris.
These matrix contaminants can interfere with the LC/MS analysis that is used to identify and quantitate the therapeutic oligonucleotide and its metabolites. Further complicating the isolation of oligos from plasma and tissue are their chemical properties. Oligonucleotides’ polar and polyanionic qualities make it difficult to use standard separation procedures to clean up plasma samples for LC/MS analysis methods.
A method for performing sample preparation using a two-step procedure of liquid–liquid extraction (LLE) and solid-phase extraction (SPE) reported in Analytical Chemistry in 2007 has met with limited success with small studies where the large amount of required manual manipulation is not a factor. However, in clinical or animal studies of a candidate therapeutic oligonucleotide, which can require the analysis of thousands of samples, this sample-prep method is not practical.
Improving Oligonucleotide Isolation
To overcome the limitations of two-step isolation, a new methodology was introduced last year for separating oligonucleotides from serum and plasma. This methodology, which has been incorporated in Phenomenex’ Clarity OTX oligonucleotide isolation kit, uses a novel solubilization buffer instead of the time-consuming LLE steps from other protocols.
This method also uses a mixed-mode SPE sorbent for specifically capturing oligonucleotides while allowing the elution of proteins and lipids that can interfere with LC/MS analysis. Being an SPE-based protocol, the isolation methodology is rapid and easily performed in less than 15 minutes.
The Clarity OTX method uses buffers that maintain near-neutral pH throughout the process to avoid unwanted modifications of the oligonucleotide (depurination for DNA below pH 5 and 2´ to 3´ isomerization for RNA above pH 8). The clean-up and recovery of this protocol is demonstrated in Figure 1.
In this example, a 27 mer DNA oligonucleotide was spiked into plasma at the 12 µg/mL level and isolated using the Clarity OTX protocol for analysis by HPLC. The upper HPLC chromatogram of Figure 1 is the plasma-extracted oligonucleotide; the lower chromatogram is the same oligonucleotide directly injected onto the HPLC system. Note the minimum number of UV-interfering contaminants in the plasma-extracted sample.
Because most matrix interference peaks elute away from the oligonucleotide, quantitation is unaffected by matrix interference. Recoveries of greater than 95% from the plasma samples demonstrate the utility of this protocol for isolating oligonucleotides from biological samples. While the data is not shown here, the new isolation method produces a linear response curve with sensitivity down to low nanomolar concentrations, depending on the LC/MS system used.
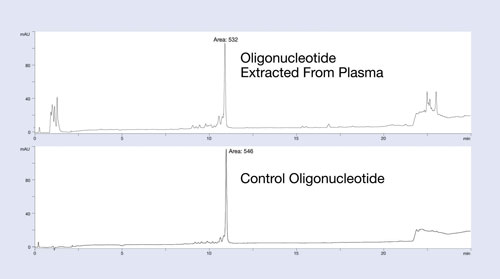
Figure 1. Recovery and extraction effectiveness studies of the isolation protocol using Clarity OTX for a 27 mer phosphorothioate oligonucleotide: The oligonucleotide was spiked into plasma, extracted using Clarity OTX, and analyzed by HPLC. The chromatogram is compared to a control sample run on the HPLC using identical conditions. Near quantitative recovery is observed.
LC/MS Analysis
In addition to isolation, the analysis of oligonucleotides by LC/MS presents unique challenges compared with the study of small molecule therapeutics. Being reasonably long anionic polymers, oligonucleotides are not generally retained by reversed-phase HPLC and require the use of ion-pairing buffers to obtain retention for analysis by LC/MS.
A good review of the balance between ion-paring mediated retention and ion-suppression is covered in detail in a May 1, 2009 article in GEN (available online). However separation of the oligonucleotide from matrix interference is an additional requirement.
An example of isolating an oligonucleotide from LC/MS-interfering matrix contaminants is shown in Figure 2. In this example a 19 mer phosphorothioate oligonucleotide was extracted from plasma using the Clarity OTX protocol and analyzed on an Oligo HTCS LC/MS system (Novatia) utilizing a Clarity Oligo-MS column.
Figure 2A shows the LC/MS TIC chromatogram of the extracted oligonucleotide and demonstrates high recovery and removal of MS-interfering plasma matrix contaminants that might co-migrate on LC. (A few matrix peaks are observed that elute away from the oligonucleotide peak.) The isolated MS spectra of the 19 mer oligonucleotide peak at 14.3 minutes retention time is shown in Figure 2B. Major ions corresponding to the -6, -7, and -8 ions of the parent oligonucleotide are readily seen.
Using an electrospray ionization mass spectrometer, oligonucleotides generally are observed as a mixture of negatively charged ions that do not readily translate to a specific parent mass of the oligonucleotide. This data is further complicated when multiple components co-migrate by LC.
To identify all of the components in an oligonucleotide mixture, deconvolution software is needed to translate signal contributions from all the different ions to the parent mass of the expected oligonucleotide as well as any major contaminants. The raw spectra from Figure 2B were input into the Novatia ProMass deconvolution software to generate the reconstructed mass spectra shown in Figure 2C.
Note that processed ions generate a mass spectrum that corresponds to the expected mass of the full-length 19 mer oligonucleotide. Also note minor components that correspond to low-level sodium adducts of the full-length oligonucleotide as well as a low-level peak corresponding to the expected mass of a depurinated oligonucleotide. While that depurinated oligo peak might possibly be due to an MS artifact, this data demonstrates the necessity of having deconvolution software for analyzing oligonucleotide samples to quantitate the oligonucleotide as well as any low-level metabolites.
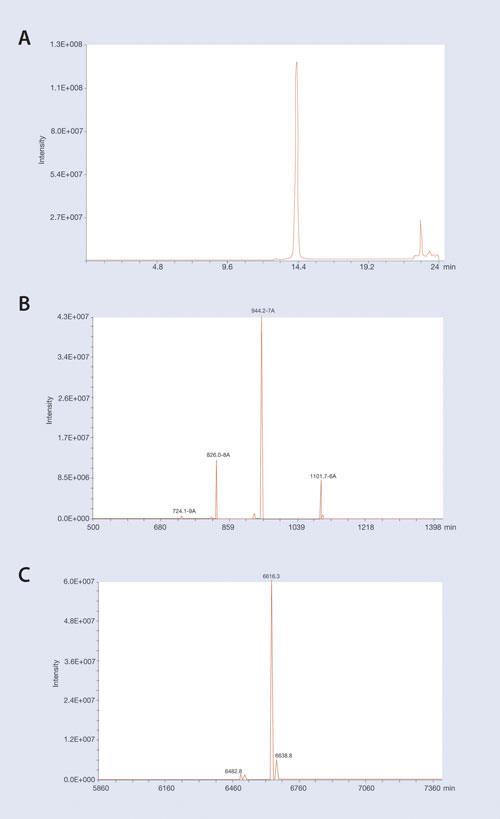
Figure 2. LC–MS chromatograms of 19 mer P-S oligonucleotide extracted from plasma using Clarity OTX: (A) The MS TIC of the extracted oligo. (B) The MS spectra at main oligo peak at 14.3 minutes. Note the majority of ions at charge states of -6 thru -8. (C) The reconstructed mass spectra generated by ProMass software give the expected parent mass at 6,616 Da. Note the low-level mass at 6,482 Da that corresponds to depurination of the oligonucleotide. Deconvolution software is required for detecting minor contaminants for Oligo MS spectra.
Summary
In the drug-development pipeline for oligonucleotide therapeutics, the most recent challenge has been developing robust, high-throughput methods for preparing and analyzing ADME/pharmacokinetic samples. The results shown here demonstrate the suitability of moving to SPE-only protocols using Clarity OTX cartridges as a way of bypassing the manually intensive LLE steps and automating the sample-preparation process.
Multiplexing the process in a 96-well plate format offers the prospect of further increasing throughput. Equally important in obtaining useful LC/MS data for oligonucleotides is using proper chromatography conditions on a high-resolution HPLC column coupled to a sensitive MS system. Finally, having proper bioinformatics to reconstruct MS spectra is crucial to quantitating oligonucleotides and their metabolites from biological samples.
Michael McGinley ([email protected]) is bioseparations product manager, Greg Scott is a research scientist, and Brian Rivera is a bioseparations specialist at Phenomenex.





