
November 15, 2011 (Vol. 31, No. 20)
Keith Hansen
ssDNA Oligos and CompoZr ZFNs More Efficiently Produce Nucleotide Insertions and/or Deletions
CompoZr® zinc finger nucleases (ZFNs) from Sigma® Life Science enable efficient and affordable gene editing across many species and cell types, including humans, rats, mice, frogs, rabbits, pigs, cattle, C. elegans, and Drosophila.
Recent literature has shown ZFNs mediating increasingly diverse types of in vitro genome modifications, some of which we describe in this tutorial, as well as the potential of in vivo genome editing through the restoration of hemostasis in a mouse model of hemophilia. Simultaneously, advances inCompoZr ZFN production and design have halved the price of both Custom ZFNs and Knockout ZFNs for any gene in the human, mouse or rat genomes, bringing CompoZr ZFNs within the budget of any laboratory.
CompoZr ZFNs work by creating a user-defined double-stranded break (DSB) that is repaired primarily by one of two natural mechanisms—nonhomologous end joining (NHEJ) or homology-directed repair (HDR). NHEJ can create nucleotide insertions or deletions (indels) that lead to frameshifts, subsequent nonsense-mediated decay, and functional gene knockout.
In the presence of a DNA donor plasmid that flanks the DSB, HDR can integrate entire transgenes, fuse reporter genes, introduce or correct a point mutation, or perform many other sophisticated genomic modifications.
Donor plasmid design, while an established process, can require several weeks from initial concept to delivery and costs hundreds of dollars. A recent Nature Methods publication by Chen et al. reports the use of ssDNA oligonucleotides (ssODNs), as opposed to donor plasmids, to derive three unique genetic outcomes—targeted point mutation, targeted gene deletion up to 100 kb using a single CompoZr ZFN, and targeted insertion of genetic elements along with large genomic deletions.
In this article, we describe the modification of a targeted codon in the human RPS6KA3 gene that encodes kinase RSK2, which is implicated in cancer, mental retardation, and psychomotor and skeletal disorders.
Methods
Oligonucleotide and ZFN engineering: A ZFN that cuts 27 bp from the site of the desired codon change in the RSK2 locus was engineered by the CompoZr ZFN operations group at Sigma Life Science. All oligonucleotides were manufactured by Sigma Life Science and did not require chemical modifications to achieve efficient genome editing.
A 125 mer RSK2 ssODN was designed that contained: (1) the codon change, a cysteine to valine switch in the ATP binding domain, (2) a silent cytosine to adenine mutation to create a BamHI restriction site to assist with genotyping during clonal isolation, (3) a ZFN blocking mutation (ZBM) to prevent re-cleaving of the modified locus post-integration of the ssODN-delivered sequence (Figure 1A).
Cell culture and transfection: The human K562 cell line was obtained from American Type Culture Collection (ATCC) and grown in Iscove’s Modified Dulbecco’s medium, supplemented with 10% FBS and 2 mM L-glutamine. K562 cultures were split one day before transfection and were at ~0.5 million cells mL-1 and nucleofected with Nucleofector Solution V (Lonza) using the T-016 program.
Each K562 nucleofection contained 500,000 cells suspended in 100 µL of Nucleofector solution. The ssODN was dissolved in 10 mM Tris (pH 7.6) at 100 µM, and 1 or 3 µL of the stock solution was mixed with 4 or 8 µg of ZFN mRNA (2 or 4 µg of each ZFN mRNA). Cells were grown at 37°C and 5% CO2 immediately after nucleofection.
Restriction fragment length polymorphism assay: To determine if ZFN editing was sufficient to begin single-cell cloning, genomic DNA was extracted from transfected cells two days after nucleofection. Genomic DNA was then PCR amplified with primers flanking the ssODN target region. For the RSK2 locus all amplicons were digested with BamHI and resolved on an acrylamide gel. The primers were designed to generate a 428 bp fragment.
Cell cloning: For the codon conversion single-cell cloning was performed by limiting dilution. Cells were lysed and screened for targeted mutations by real-time PCR. Clones were screened with a BamHI site-specific forward primer and a wild-type specific forward primer in combination with a common reverse primer. Candidate clones were then screened for the biallelic cysteine to valine mutation with a mutation-specific forward primer and a wild-type specific forward primer in combination with the common reverse primer.

Figure 1. (A) A 125 mer oligo-donor designed to incorporate three desired mutations in the RSK2 locus. (B) Acrylamide gel separation of BamHI-digested K562 genomic DNA two days after transfection of oligos and ZFNs. The intensity of the BamHI bands relative to the parental PCR band is a measure of the mutation frequency in the pool of cells.
Results
Using the RSK2 125 mer ssODN and RSK2 ZFNs, BamHI conversion rates of 22–32% were detected by the RFLP assay (Figure 1B). Single-cell cloning yielded at least 20 clones from 750 with a biallelic BamHI conversion (Figure 2A) and the desired cysteine to valine codon modification (DNA sequencing data not shown). Immunoblotting of three randomly selected mutants demonstrated the predicted insensitivity of RSK2 to the RSK kinase inhibitor fmk (Figure 2B).
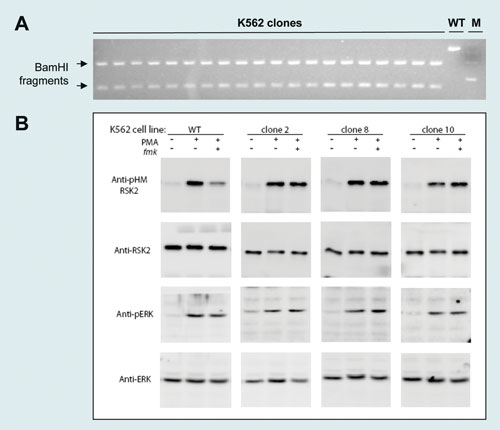
Figure 2. (A) 3% agarose gel displaying desired biallelic BamHI mutation in 20 clones. (B) Immunoblots probing the sensitivity of wild-type and RSK2-C436V modified cells to the small molecule RSK kinase inhibitor fmk. Three independent human K562 clones were tested.
Discussion
As a method for characterization of eukaryotic proteins, codon-based mutagenesis has been restricted to cDNAs harbored on exogenous plasmids. In this application, the coding sequence for the RSK2 kinase was modified at the endogenous location on human chromosome X in the context of: (1) the native promoter and all associated 5´-UTR regulatory elements, (2) intron/exon splicing, (3) mRNA export and processing, and (4) any 3´-UTR regulatory elements such as microRNA binding sites. This ensures that the expression levels of the RSK2 kinase and associated small molecule kinetics are controlled in the truest biological fashion.
Previously, this mode of endogenous protein manipulation was limited to certain model bacteria and yeast. Now, it is a realistic experimental option for mammalian cells using ZFNs in combination with ssODN or plasmid donors.
A major technical challenge for ZFN-based genome editing has been the engineering of ZFNs within close range (<200 bp) of the desired mutation site. The use of ssODNs layers an even tighter restriction on ZFN design resolution since ssODN length is often limited to <130 bp for economical and quality reasons.
Any genome-editing method must achieve a high level of accuracy to be useful. CompoZr ZFNs utilize a multifaceted approach that includes bioinformatics, FokI nuclease engineering, and a proprietary library of ZFN modules to assure specificity. Bioinformatics is used to avoid homologous genomic regions, SNPs, splice variants, and repeat elements.
Also, nearly all CompoZr ZFNs have target sites at least 30 bp long that differ from any other site in the genome of interest by at least four nucleotides. These bioinformatic steps assure specificity and are applied to all CompoZr-engineered ZFNs, both Custom ZFNs and readily available human, rat, or mouse gene Knockout ZFNs.
In addition to these strict bioinformatic measures, CompoZr ZFN’s FokI domain requires heterodimerization to create a DSB, providing a significantly higher level of specificity for all CompoZr ZFNs regardless of the genetic locus being studied.
Combining the use of ssODN templates and exacting measures to ensure the specificity of CompoZr ZFNs maximizes the efficiency and subsequent utility of CompoZr ZFN-mediated genome editing.
Keith Hansen ([email protected]) is product manager of Emerging Technologies at Sigma Life Science.





